









by Alexander Schönhuth (CWI and Utrecht University) and Leen Stougie (CWI and VU Amsterdam)
Many life-threatening viruses populate their hosts with a cocktail of different strains, which may mutate insanely fast, protecting the virus from human immune response or medical treatment. Researchers at CWI have designed a method, named Virus-VariationGraph (Virus-VG) [3], that puts all strains onto a graphical map, which facilitates more reliable and convenient identification of potentially resistance-inducing or particularly lethal strains.
Viruses, such as HIV, Ebola and Zika, populate their hosts as a viral quasispecies: a collection of genetically related mutant strains, which rapidly evolve by the accumulation of ever more mutations as well as recombination among the strains. To determine the right treatment for infected people, it is crucial to draw a clear picture of the virus DNA that affects the patients [1]. The genome of an HIV strain, for example, consists of approximately 10,000 letters. While most virus strains generally share most letters, comparatively rare, but utterly relevant differences can decisively determine their clinically relevant properties, such as resistance to treatment, or their virulence. To draw a clear picture, it is necessary to, first, reconstruct the genomes of the different strains at full length, and second, to estimate the relative proportions of the strains that make up the viral quasispecies, the mix of strains affecting an individual patient.
Applying modern sequencing techniques to virus DNA extracted from infected people yields millions of sequence fragments, however, and not full-length genomes of strains. The task is now to assign the (many) fragments to different strains. Each genome then needs to be reconstructed at full length, and its relative abundance estimated within the mix of strain genomes. This procedure is commonly referred to as viral quasispecies assembly. It is important to note that virus reference genomes, which seem to promise orientation during the assembly process can considerably disturb this procedure, by introducing biases that can decisively hamper the assembly.
Viral quasispecies assembly is very challenging, particularly in the absence of reference genomes, and is not yet a fully resolved issue. Schönhuth, Stougie and their co-workers have recently taken big strides in this area.
Their idea was to put all fragments (or better: contigs, which are contiguous patches of fragments that together must stem from an identical strain; these can be reliably determined using other methods [2]) on a directed, graphical map. In such a map, full-length paths correspond to full-length genomes. Further, the relative abundance of a strain genome then relates to the relative number of fragments that make part of the path through this map. This graphical map then allows low-frequency strains - paths through the map that are supported by rather low amounts of fragments - to be conveniently highlighted. The identification of low-frequency strains is important in the analysis of viral quasispecies. When not subjected to a careful analysis, low-frequency strains tend to be neglected, and consequently such strains may induce resistance to treatment or emerge as particularly virulent after treatment.
Schönhuth, Stougie and co-workers have developed a method, Virus-VariationGraph (Virus-VG) that implements these ideas. This was achieved through the construction of “variation graphs” from the input fragments (which are contigs, see above). Variation graphs have become popular recently in the analysis of genomes. The general idea is to transform a collection of related genomes into a variation graph, which allows for types of genome analyses that were hitherto unconceivable. Usually, however, variation graphs are constructed from full-length genomes, which prevents the use of variation graphs for viral quasispecies assembly.
Here, Schönhuth, Stougie and co-workers generalised the concept of variation graphs, which allowed them to be flexibly constructed from shorter sequence patches. They designed an optimisation problem whose solution consists of laying out the paths that correspond to strain genomes, and assigns relative abundances to those paths. See Figure 1 for an illustration of the steps.
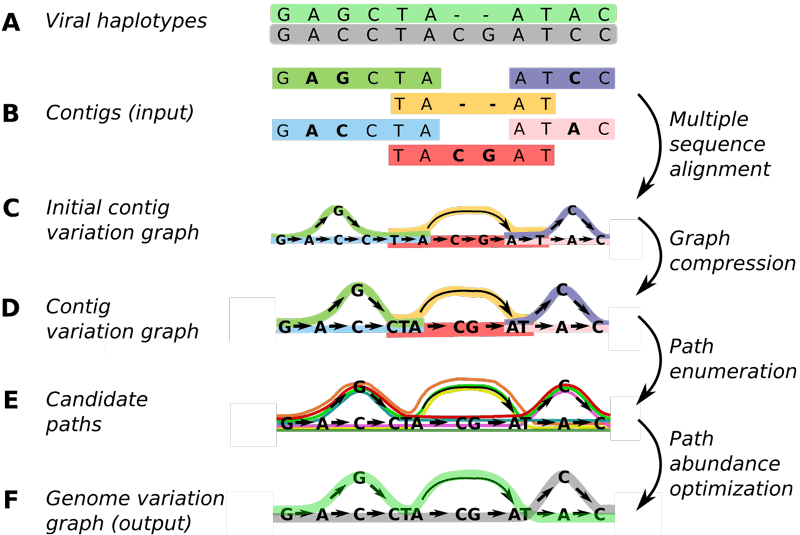
Figure 1: At CWI, researchers have developed Virus-VG, an algorithm that is more reliable and convenient to use for assembling viral quasispecies than earlier methods. Picture: CWI.
They were able to demonstrate the advantages of the new graph-based approach over other viral quasispecies approaches (all of which use reference genomes), in various relevant aspects, such as strain coverage, length of genomes, and abundance estimates. This method seems especially beneficial for identifying low-frequency strains, which is of particular interest for the above-mentioned clinical reasons.
Overall, Schönhuth, Stougie and co-workers succeeded in providing the first solution to the viral quasispecies assembly problem that does not only yield the genomes of the strains at maximal length, but also reliably estimates their relative abundances, without making use of existing reference genomes. Virus-VG is publicly available at [L1].
Link:
[L1] https://kwz.me/hy3
References:
[1] S. Posada-Cespedes, D. Seifert and N. Beerenwinkel: “Recent advances in inferring viral diversity from high-throughput sequencing data”, Virus research, 239, 17-32, 2017.
[2] J. Baaijens, et al.: “Full-length de novo viral quasispecies assembly through variation graph construction”, Bioinformatics, btz443, https://doi.org/10.1093/bioinformatics/btz443, 2019
[3] J. Baaijens, A.Z. El Aabidine, E. Rivals, A. Schönhuth: "De novo assembly of viral quasispecies using overlap graphs", Genome Research, 27(5), 835-848, 2017.
Please contact:
Alexander Schönhuth.
CWI, Netherlands.

