









by Karl N. Kirschner, Axel Arnold, and Astrid Maaß
Multiscale modelling requires the transferral of knowledge gained at different resolutions. Through the use of an expert-driven workflow we have developed reliable pathways for transferring information from quantum mechanics to atomistic and coarse-grained simulations.
The research field of computational chemistry had its beginnings in the early 1900s. However, its widespread use wasn't realized until the last half of the century, coinciding with significant increases in computer power and algorithm sophistication. A useful way to categorize the field is by considering resolution, in terms of both particle size and molecular event time scale. For example, examining the behaviour of electrons interacting with nuclei is best accomplished using quantum mechanics (QM) calculations, while understanding the binding of a ligand to a biological receptor is easily done using atomistic molecular dynamics (AT-MD) simulations. In the last two decades there has been an increased interest in studying systems at multiple resolutions, which is commonly known as multiscale modelling. One of the strengths of multiscale modelling is its ability to provide and link a system's physics at different time scales, thus providing a unique understanding that is otherwise difficult to obtain.
One of our goals is to model chemical and biological systems at different levels of resolution, ranging from QM calculations to coarse-grained MD (CG-MD) simulations as illustrated by Figure 1. A challenging task for all multiscale modelling is to transfer the knowledge gained from one resolution to another. As such, there is an ongoing need for the development of knowledge transfer procedures.
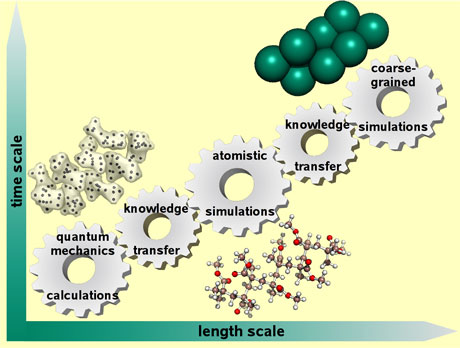
Figure 1: A representative scheme for multiscale molecular modelling. Poly(methyl methacrylate) polymer, or Plexiglas, is shown as a representative molecule.
Consider, for example, our desire to model lipid layers. QM calculations give us insight into the molecular potential energy surface of small lipids. We can also divide lipids into different chemical functionalities (eg hydrocarbons, alcohols) that can be explored using QM calculations, thus providing a better understanding of how these functionalities are coupled within the parent molecule. The resulting knowledge can be transferred to AT-MD simulations through force-field parameterization.
To achieve our goals, we have developed an expert-driven workflow package that allows parameter development to be partly automated. Such a workflow allows the researcher to influence how knowledge proceeds, ensuring that insights gained from QM are effectively transferred to atomistic simulations. The following is a brief outline of the workflow:
1. Reliable QM geometry optimizations are performed, constraining the degree of freedom of interest.
2. A single self-consistent field calculation is performed at a more rigorous QM level (eg including electron correlation) on each optimized geometry, providing more reliable relative energetics.
3. Selected conformations are chosen based on the molecule's potential energy surface; subsequently, additional QM calculations are performed to determine appropriate partial atomic charges.
4. Atom types are assigned to each atom. Molecular mechanics (MM) optimizations are performed, constraining the same degree of freedom as in Step 1.
5. The parameter that corresponds to this degree of freedom is optimized through comparison of the QM and MM relative potential energy curves (ie Steps 1 & 4).
The researcher influences the workflow by deciding, in part, which small molecules are relevant for parameterization (eg alcohols for terms involving the hydroxyl group), which QM levels are appropriate, which conformations are most relevant for computing partial atomic charges, and which conformations are preferentially weighted during parameter optimization. The transfer quality of the QM-gained knowledge to AT-MD simulations ultimately resides in the expertise of the researcher performing the transfer.
Using our workflow package, we have developed a reliable force field called Explicit Torsion Parameters (ExTrM), which uses QM data for parameter optimization and experimental data validation. Our goals for ExTrM are that (i) it is transferable between molecular systems; (ii) only explicit parameter terms are used; (iii) the number of atom types is kept to a minimum, (iv) the parameters are kept as small and close to a chemical meaning as possible; (v) 1-4 nonbonded and electrostatic scaling factors are unused; and (vi) gas- and solution-phase properties can be reproduced. Currently, we have performed over 6000 QM calculations to optimize nearly 200 parameters, covering a wide range of chemical functionalities. We have also shown reliable transferability and reproduction of gas- and solution-phase properties for selected small molecules.
In general, once the parameters have been optimized and validated, they are ready for implementation in an AT-MD simulation. Once finished, the atomistic knowledge gained can be transferred to a CG model. This may be accomplished by optimizing a CG potential to reproduce AT-MD observables (eg radial distribution curves). In collaboration with the Max Planck Institute for Polymer Research, located in Mainz (Germany), we are developing the Extensible Simulation Package for Research on Soft matter (ESPResSo++), which is specifically designed for simulating CG models, as well as the necessary tools to transfer the atomistic-gained knowledge to this resolution.
Using the above techniques, we are poised to deliver molecular insight at different resolutions for many molecular and biological systems. Such systems include, but are not limited to, organic compounds, ionic liquids, solvents, drug-like compounds, carbohydrates and lipids. Possible insights include important molecular interactions, thermodynamic properties, density, diffusion rates and toxicity.
Links:
Fraunhofer Institute for Algorithms and Scientific Computing (SCAI):
http://www.scai.fraunhofer.de/en
Max Planck Institute for Polymer Research:
http://www.mpip-mainz.mpg.de
ESPResSo++:
http://www.espresso-pp.de
Please contact:
Karl N. Kirschner
Fraunhofer SCAI, Germany
Tel: +49 2241 14 2052
E-mail:
