









by Alina Sîrbu, Heather J. Ruskin and Martin Crane
Integration of large amounts of experimental data and previous knowledge is recognized as the next step in enhancing biological pathway discovery. Here, data integration for quantitative regulatory network modelling is under investigation, using evolutionary computation and high-performance computing.
With the completion of the genome project and advances in high-throughput measuring techniques, a base for systems biology research has been created. This involves uncovering biological pathways and networks between cell products, and is an important step in finding disease markers and treatments, and in the future, toward building synthetic organisms.
Such aspirations have triggered considerable research, spanning multiple fields such as mathematics, computer science and biology, resulting in enormous amounts of data describing cellular processes, and stimulating alternative modelling efforts. However, most of these data are diverse and largely unreconciled, so that modelling approaches can offer only limited insight. Thus, in recent years, integrative approaches to modelling biological networks have appeared. However, the number of data types is usually small compared to the potential set, so that biological realism is only approximate.
The Centre for Scientific Computing and Complex Systems Modelling (Sci-Sym) was formally established at Dublin City University in 2007. It links existing research groups in computing, such as ModSci (Modelling and Scientific Computing) and mathematics. It conducts multi-disciplinary research in complex systems modelling, ranging from biological to socioeconomic systems. One of the centre's recent projects has been to build qualitative models for gene regulatory networks (GRNs). This was funded by the Irish Research Council for Science, Engineering and Technology.
GRNs consist of interactions between proteins, known as transcription factors, and genes, which in turn encode other proteins. Transcription factors bind to a DNA region close to the target gene and activate or repress its expression, ie the formation of the encoded protein. This creates complex networks of activation and repression links which, by controlling protein levels in cells, are involved in important processes such as cell differentiation, cell cycle or response to external shock.
Two types of models have been applied to GRNs: coarse-grained, which allow for large-scale low-resolution analysis, and fine-grained, for low-scale high-resolution studies. The latter enable continuous simulation of gene expression, and are, from this point of view, important tools in predicting outcomes of different perturbations in the network, corresponding to diseases or treatments. However top-down models have the disadvantage of size limitation, with only small networks feasible for this approach and the computational power currently available. In this context, a principal aim is to increase the scale capability and quality of quantitative models, in order to perform reliable simulations of entire GRNs. From a software engineering point of view, the objective is to obtain a user-friendly software application, which handles the computational and mathematical aspects of modelling, to enable biologists to focus on validation and interpretation of results.
To achieve this objective, one solution is to adopt an integrative approach. Given the large amount of available data on gene expression dynamics in different biological processes, we investigate means to integrate these to increase the scale capabilities of quantitative gene expression modelling, using evolutionary computation. Evolutionary algorithms are known to perform very well in large search spaces and with limited data, making them well suited to this problem. They have the advantage of flexibility in terms of adding different types of data to the inferential process. Furthermore, they are intrinsically parallel, so multi-threaded implementations are straightforward, facilitating the use of the local (Sci-Sym) high-performance computing cluster, and subsequent extensions to the Irish Centre for High-End Computing facilities.
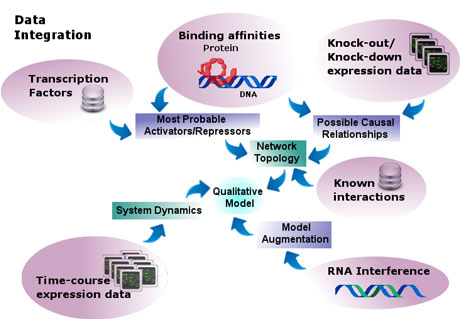
Figure 1: Data integration.
Typically, dynamical models of GRNs are inferred using time-course gene expression data. However, due to experimental costs, time series are usually very short, containing less than 100 measurements, while networks can be very large, involving hundreds of genes. There do exist multiple time series from different sources describing the same process, but they are measured on heterogeneous platforms, so analysis is not straightforward. To our knowledge, integration of these time series in the context of GRN qualitative modelling has not previously been attempted, and requires considerable pre-processing. We have analysed integrated gene expression data coming from three microarray platforms in preliminary studies, using different pre-processing methods to provide a comparative framework and the basis for a single model. Statistical integrity is a major consideration. Nevertheless, we have shown that differential equation models built from multiple datasets, are more robust to parameter and data perturbations, and display less noise overfitting. This provides the first step towards improving qualitative models for regulation.
The integration process will continue with knock-out and knock-down measurements of gene expression, introduced at the initialization stage of the evolutionary algorithm. Information on known transcription factors and interactions will be used to implement customized genetic operators, driving the inferential algorithm towards richer areas in the search space. Binding-site information will also be included in model fitness evaluation, to reward models that connect genes and proteins with high affinities. Finally, we plan to introduce RNA interference in the modelling process, first as metadata, then as distinct nodes in the regulatory network.
Links:
http://sci-sym.computing.dcu.ie/
http://www.computing.dcu.ie/~msc/
http://wiki.c2b2.columbia.edu/dream/index.php/The_DREAM_Project
Please contact:
Alina Sîrbu
Dublin City University, Ireland
Tel: +353 1 700 6747
E-mail:
