









by Filippo Geraci and Marco Pellegrini
Advanced personalized medicine is one of the goals of current research, and in this area new genetic diagnostic methods are critical. Diagnostic technology that can be used in the field (closer to the patient, and far from the traditional high-tech labs) is needed. We are studying this problem with respect to technology for personalized chromosome haplotyping.
The Human Genome project (1990-2003) highlighted the fact that humans share over 99% of their DNA. In the last few years however, the attention of researchers has shifted from what is common among members of the same species (the genome), to what is different. The single nucleotide polymorphisms (SNPs, pronounced ‘snips’) are the most common form of variation in human DNA. The total number of SNPs present in the human genome is estimated to be about 9-10 million and currently about 3.1 million have been profiled. The set of SNPs present in a chromosome (called the haplotype) is of interest in a wide area of applications in molecular biology and biomedicine. Personalized haplotyping of (portions of/all) the chromosomes of individuals is one of the most promising basic ingredients leading to effective personalized medicine (including diagnosis and therapy).
Luckily, personalized haplotyping is now becoming feasible due to a steady decrease in the cost of sequencing equipment that will soon render cost-effective the effort of producing complete genomic profiles of individuals versus collecting data on predefined genetic markers. The first publication of a complete individual diploid human genome sequence appeared in 2007. The current aim of the research community is to be able to sequence individual genomes at a cost of just 1000 USD within a few years. The actual trend in the cost of state-of-the art technology suggests that this target is realistic.
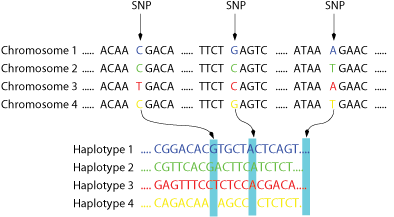 Figure 1: A family of four chromosomes in which the SNPs are highlighted is shown on top.
Figure 1: A family of four chromosomes in which the SNPs are highlighted is shown on top.
Below the corresponding haplotypes.
A key component of the reconstruction pipeline is the so-called ‘Single Individual Haplotyping’ problem (SIH). PCR-based shotgun technology produces a large collection of overlapping fragments, each containing a few SNPs. By using reference maps it is relatively easy to locate the position and orientation of such fragments in the chromosome, but not to determine the association of the fragments with the two homologous copies (paternal and maternal) of a chromosome. From the biological point of view, the single individual haplotyping problem consists in determining the association of each fragment with one of the two homologous chromosomes and then determining the haplotype.
From the computational point of view the problem was first formalized by Giuseppe Lancia in 2001. It can be described as follows: each fragment is represented as a string of fixed length such that each character corresponds to a nucleotide or to a gap (if the fragment does not cover a certain position). Each SNP can be covered by a certain number of fragments and can take only two values (the values of the haplotype in that position). The natural way of representing fragments is to store them in a matrix (the SNP matrix). We say that two fragments (rows of the SNP matrix) are in collision if they have a different value for at least one SNP in a certain position (not a gap).
Given the SNP matrix, we can define the conflict graph as follows: for each fragment there is a vertex; if two fragments are in collision, insert an edge between the corresponding vertices. When the SNP matrix contains no errors, conflicts can only arise between fragments originating from different haplotypes; therefore, the corresponding conflict graph is bipartite.
In the absence of errors or gaps, with sufficiently high coverage, this problem is easily solved. However, more realistic models of the problem take into account several types and sources of error/noise in the data, and in this setting the problem is more challenging (intractable in some cases). Several algorithms and heuristics have been proposed in the literature to solve the haplotype assembly problem in this more realistic setting.
A common feature of these algorithms is that, due to lack of a common benchmark and of publicly available implementations in a unified framework, experimental comparisons between different methods are based on a narrow choice of data, parameters and algorithms. So far, in large scale haplotyping projects, insufficient attention has been given to the problem of how to identify the most suitable algorithm for a specific experiment and/or technology. To remedy this situation, and to provide a service to the bioinformatics community, we have developed the easy-to-use Web-based tool ReHap for testing diverse algorithms, on a variety of data sets, and with a variety of parameter settings.
Within ReHap our research group has developed a novel and fast heuristic method for the SIH problem called SpeedHap, which handles gaps efficiently, and is able to deal with high reading error rates (up to 20%) and low fragment coverage (as low as 3): situations in which other methods fail. We demonstrate these properties via experiments on real human data from the HapMap project. Away from the controlled environment of a lab it is likely that the current portable technology for sequencing will produce less reliable data. Moreover, if a real-time and high throughput response is needed to care for the needs of many individuals in a short time span, one might not be able to guarantee a high coverage of the fragments and low reading error rates. SpeedHap is a step towards the efficient extraction of useful information even from low quality data.
Rehap and SpeedHap have been developed by the Bioalgo Group at the Institute for Informatics and Telematics of CNR.
Link:
http://bioalgo.iit.cnr.it/rehap
Please contact:
Filippo Geraci
IIT-CNR, Italy
E-mail:
