









by Gianna Serafina Monti (University of Milano-Bicocca), and Peter Filzmoser (TU Wien)
E-values provide a principled foundation for false discovery rate control in high-dimensional microbiome data analysis. We show how their aggregation properties enable a derandomized, robust knockoff filter that outperforms classical approaches in stability and reproducibility.
Identifying which bacterial species in the gut microbiome are genuinely associated with clinical outcomes, such as obesity, liver disease, or cancer, is a central challenge in modern computational biology. The statistical hurdles are formidable: microbiome data are compositional (only relative abundances are observed), high-dimensional (far more species than subjects), sparse (few species are truly related to outcomes), and frequently contaminated by outliers. Clinical studies also routinely measure several outcomes simultaneously, calling for multivariate methods rather than repeated univariate analyses.
A recurring challenge in this setting is controlling the false discovery rate (FDR), the proportion of spurious associations among those reported, while maintaining adequate power to detect true signals. P-value-based FDR control often relies on restrictive assumptions about the data’s dependence structure – assumptions that are frequently violated in microbiome studies – whereas e-values provide a more flexible framework. Their natural closure property under averaging enables robust error control even in high-dimensional settings with complex, unknown dependencies. An e-value is a non-negative number that quantifies evidence against a null hypothesis: a value of 1 indicates no evidence, while large values indicate strong evidence. Unlike p-values, e-values can be freely averaged and combined across independent experiments without compromising their validity as evidence measures. This closure property under averaging is the key to a new generation of derandomized procedures: by aggregating e-values across repeated knockoff constructions, we achieve a level of selection stability and reproducibility previously unattainable with p-value-based filters.
The knockoff framework and its randomness problem
The knockoff filter is a powerful approach to variable selection with FDR control. It works by augmenting the dataset with synthetic copies of the predictors, the “knockoffs”, that mimic the correlation structure of the originals but carry no signal. Comparing the regression coefficients of the original and knockoff features yields evidence scores, and a data-driven threshold selects features above a level that provably controls the FDR. The Multi-Response Knockoff Filter (MRKF) of Srinivasan et al. (2023) extends this idea to multivariate response settings, making it applicable to studies with multiple clinical outcomes.
However, the classical knockoff filter has a well-known Achilles’ heel: its inherent randomness. Because the knockoff matrix is inherently stochastic, repeated analyses of the same dataset yield different sets of selected variables – an undesirable property in scientific practice, where reproducibility is paramount. This instability is not a minor inconvenience; it can undermine trust in reported findings and complicate downstream biological interpretation.
E-values as the bridge to derandomization
We show that e-values provide an elegant and principled solution to this instability [2]. The key idea is to run the knockoff filter M times, each with an independently sampled knockoff matrix, and to convert each run’s selection into e-values for each feature. Specifically, for each feature and each knockoff copy, an e-value is defined that is large when the feature is selected and small otherwise. Because these e-values are independent across runs, their average is also a valid e-value – a direct consequence of the martingale structure underlying e-value theory. The averaged e-values are then fed into the e-BH procedure [3], which controls the FDR under arbitrary dependence among hypotheses. The result is a single, stable selection set that aggregates evidence across multiple randomizations without inflating the FDR.
This derandomized procedure, which we call RobMRKF-Derand, inherits all the finite-sample FDR guarantees of the knockoff framework while dramatically reducing variability across runs. The e-value aggregation step is not merely a computational trick. It reflects a genuine accumulation of statistical evidence, consistent with the broader e-value literature on sequential and anytime-valid inference.
Robustness and compositionality
We embed this derandomized knockoff filter within a robust multivariate regression framework for compositional covariates. As the microbiome predictors are compositions and thus live in the simplex sample space, they are first mapped to the Euclidean space via the additive log-ratio (alr) transformation. Regression and covariance estimation are then performed jointly using an adaptation of the robust multivariate lasso with covariance estimation from Chang and Welsh (2023) [1], replacing the standard squared loss with Tukey’s biweight loss to down-weight outlying observations. The combination of robust estimation and e-value-based derandomization yields a method that is both resistant to contamination and stable across repeated analyses.
Results
Simulation studies confirm that RobMRKF-Derand controls the FDR at the nominal level across a range of contamination scenarios while achieving higher power and substantially lower selection variability than the non-robust MRKF. In real microbiome data applications, the method identifies biologically interpretable associations between microbial species and clinical parameters, findings that are reproducible across runs, unlike those of the classical knockoff filter.
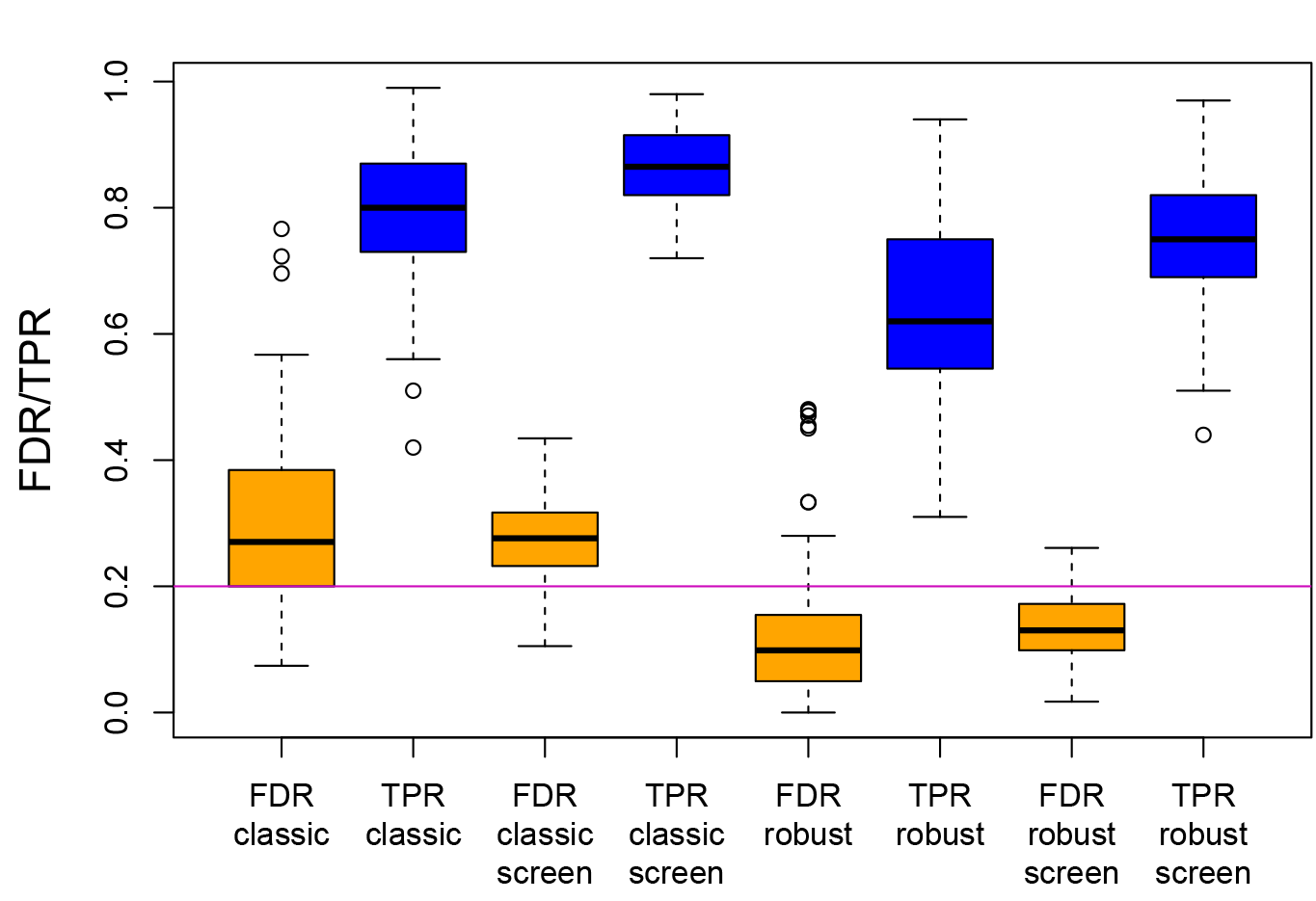
The figure presents simulation results for the FDR and TPR (true positive rate) when comparing the non-robust (classical) and robust procedures, with and without an initial screening step. In contrast to the classical method, the robust procedure keeps the nominal FDR level of 0.2 in most cases (for details, see [2]).
Links:
[L1] R code, synthetic data illustrations, and documentation: https://github.com/giannamonti/RobMReg.
References:
[1] L. Chang L, A. H. Welsh “Robust multivariate lasso regression with covariance estimation”. J Comput Graph Stat, 32, 2023
[2] G.S. Monti, M. Pujolassos, M. Calle Rosingana, P. Filzmoser, “Robust multivariate regression controlling false discoveries for microbiome data”, Bioinformatics, 41, 9, 2025
[3] Z. Ren, R. Barber, “Derandomized knockoffs: leveraging e-values for false discovery rate control,” J. R. Stat. Soc. B, 86, 2024
Please contact:
Gianna Serafina Monti
University of Milano-Bicocca

