








by Dirk Reith and Karl Kirschner
Computational chemistry began with the birth of computers in the mid 1900s, and its growth has been directly coupled to the technological advances made in computer science and high-performance computing. A popular goal within the field, be it Newtonian or quantum based methods, is the accurate modelling of physical forces and energetics through mathematics and algorithm design. Through reliable modelling of the underlying forces, molecular simulations frequently provide atomistic insights into macroscopic experimental observations.
There is great demand for well coded software that incorporates new technology on a regular basis. Since increasing numbers of atoms are needed to simulate increasingly complex systems, computing resources can easily be a limiting factor for scientists in this field, hence there is a real need for efficient software. Although there are corporate-based developments, most software packages are written by natural scientists rather than software engineers. This is due to the demanding physical and chemical concepts that need to be transferred into proper algorithmic solutions. Both academic and open-sourced development, plus some corporate software, tend to be POSIX command-line based, whose input and output can be easily parsed by researchers. We exploit this command line and parsing ability in designing new software tools that are tailored to specific research needs. Our goal is to take advantage of the strengths of various existing third-party software by creating links between them, and subsequently develop new algorithms that allow us to incorporate state-of-the-art ideas. This demands that our software should evolve to a) communicate with new releases of third-party software and b) to incorporate new ideas presented in the primary research literature.
Our solution for enabling our software to evolve is to decouple tasks within the software. In doing so, algorithmic solutions can be introduced in a modular fashion, allowing us to easily identify and update specific tasks as needed. Using these ideas, our group has developed two independent software tools, called WOLF2PACK and GROW. A third tool called ESPResSo++ has been developed in collaboration with the Max Planck Institute for Polymer Research (MPI-P) in Mainz. Taken together, these tools enable us to quickly investigate a diversity of chemical and biochemical problems. Figure 1 shows that they serve on different, but inter-linked, resolutions of molecular modelling. A brief summary of each program follows.
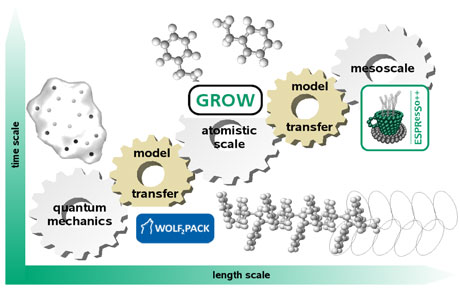
Figure 1: Current evolving software packages (co-)developed at Fraunhofer SCAI and how they fit into the multiscale modelling approach of computational chemistry.
The quality of results from molecular simulations is heavily dependent upon the quality of the underlying molecular parameters. While very important, this requires specialized knowledge and is very time consuming. Worse, the procedure differs for intramolecular coordinates (which can be derived from quantum mechanics(QM)) in comparison to the intermolecular interactions (which can only be matched versus macroscopic observables as measured by experiments). Therefore, one of the primary goals of our research is to develop accurate and reliable molecular parameters in a reasonable time and as error free as possible.
For intramolecular interactions, we have created a scientific “Workflow for force-field optimization package” (WOLF2PACK) that incorporates our beliefs for how QM-gained knowledge should be transferred to Newtonian-based models. We define a scientific workflow as a series of independent steps that are linked together according to the data flow and the dependencies between them. For intermolecular interactions we developed a systematic optimization workflow, based on efficient gradient-based numerical algorithms called GROW. GROW is a modular tool kit of programs and scripts. It is a generic implementation and can be easily extended by developers. Both tools are written by natural scientists and software engineers to really make mathematics meet chemistry. They facilitate the a) development and optimization of molecular parameters for a given simulation engine, b) transfer of parameters from one software package to another, and c) testing of the parameters using a standard test suite and protocol via an semi-automated iterative parameterization process.
Thirdly, the Extensible Simulation Package for Research on Soft matter systems (ESPResSo++), jointly developed with the MPI-P, is a parallelized, object-oriented simulation package designed to perform many-particle simulations of molecular chemical systems. The workflow idea is realized in ESPResSo++, firstly, in its model builder that allows scientists to easily create chemical systems in a flexible manner. Secondly, the modular design is created along the physics of the systems, allowing for the efficient addition of new physical effects through algorithms. The realization of both aspects is also aided by the separation of the algorithmic kernel and a simulation design and control front end. The ESPResSo++ kernel ensures efficiency through the use of advanced C++ programming language features, high-performance storage techniques, and cache optimization while users steer and control simulations by a Python script. This approach makes it easy to conduct online analysis and interactive simulations. The program structure should enable scientists to use ESPResSo++ as a research platform for their own methodological developments, which at the same time allows the software to grow and acquire the most modern methods.
Taken together, the joint efforts of natural scientists and software engineers greatly enhance software development for molecular modelling. The concept of evolving software seeded into the code development itself is a key for maintaining state-of-the-art tools and research.
Links:
http://www.scai.fraunhofer.de/coche.html
http://www.espresso-pp.de
Please contact:
Dirk Reith
Fraunhofer Institute for Algorithms and Scientific Computing (SCAI), Germany
Tel: +49 (0) 2241 14 2746
E-mail: