









by Catia M. Machado, Francisco Couto, Alexandra R. Fernandes, Susana Santos, Nuno Cardim and Ana T. Freitas
Hypertrophic cardiomyopathy is a disease characterized by a high genetic heterogeneity with variable clinical presentation, thus rendering the possibility of personalized treatments highly desirable. This can be achieved through the integration of genomic and clinical data with Semantic Web technologies, combined with the identification of correlations between the data elements using data mining techniques.
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic disease that may afflict as many as one in 500 individuals, being the most frequent cause of sudden death among apparently healthy young people and athletes. It is an important risk factor for heart failure disability at any age and is characterized by a hypertrophied, non-dilated left ventricle, myocyte disarray and interstitial fibrosis.
Since the disease is characterized by a variable clinical presentation and onset, its clinical diagnosis is difficult prior to the development of severe or even fatal symptoms. Therefore, its early diagnosis is extremely important.
In terms of genetic manifestation, more than 640 mutations in more than 20 genes have been associated with HCM phenotype. The detection of these mutations in the genome of the patient can greatly improve the disease diagnosis. However, genetic diagnosis using dideoxi-sequencing techniques is hampered by these high numbers of genes/mutations and by the fact that some patients present the clinical manifestations of the disease but none of the mutations is found under the current genetic testing (which indicates that even more mutations and/or genes might be involved). Moreover, HCM’s severity may not be the same for two individuals, even if direct relatives, since the presence of a given mutation can have a benign pattern in one individual and result in sudden cardiac death in another.
These complicating factors indicate that a joint strategy among clinicians, biologists and bioinformaticians may be advantageous. Biologists need to identify and characterize new mutations with high throughput techniques, thus enabling clinicians to count on this information, when articulated with the clinical findings, to provide the best possible treatment for each individual patient. Bioinformaticians have the tools needed to expeditiously integrate and analyse the data, thus providing the necessary articulation between the different types of data and the identification of patterns that might shed light into the disease variability.
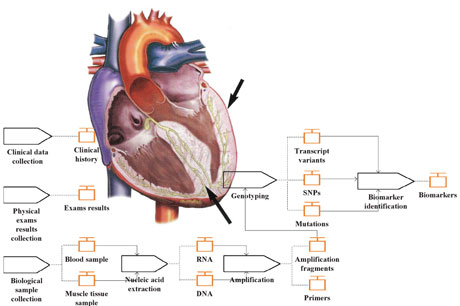
Figure 1: Model of a human heart showing the thickening of the left ventricular wall, and the HCM characterization workflow, comprising all modelling activities.
In more concrete terms, the data elements that need to be integrated correspond to the presence/absence of each mutation in the genome of the patients (genotype data) and to the clinical elements upon which the clinicians rely to provide a diagnose (phenotype data). The latter normally include the results from physical examinations (eg electrocardiogram, echocardiogram), as well as the clinical history of the individual (eg age at diagnosis, sudden deaths in the family).
The data integration procedure in this context is a very demanding task considering that genotype and phenotype data correspond to transversal domains, normally stored under heterogeneous formats and on different physical locations. The approach proposed here is based on Semantic Web technologies, previously identified as suitable for this type of task since they make it possible to integrate, share and reuse data in an application- and domain-independent manner.
Upon completion of the data integration phase, the data will be analysed with data mining techniques in order to infer genotype-phenotype correlations, or, more specifically, to develop models for the association between the presence of certain mutations and the resulting physical traits. Although a large number of studies have been conducted on the linkage between specific mutations and the risk of specific illnesses (eg cystic fibrosis), models for the more general case of genotype to phenotype association in the presence of high disease complexity, both genetic and clinical, remains largely unexplored. Supervised machine learning techniques, such as decision trees and support vector machines, offer the potential to identify more complex relationships than those identified using simple correlation analysis, the standard practice in genotype-phenotype association models. Standard statistical analysis may identify correlations between one or a small set of specific mutations, but in more complex cases these correlations will not be significant enough to lead to concrete diagnosis methods. The models obtained using data mining techniques are expected to be of great interest both in terms of their predictive ability and their practical usability for doctors.
The ultimate goal of this work is to develop a clinical characterization system that, upon introduction of a new patient’s data, will provide an indication of whether the patient is suffering from HCM based on the existing model.
Currently in the integration stage, this work is being conducted in the LASIGE group at the Departamento de Informática of the Universidade de Lisboa and in the KDBIO group at the Instituto de Engenharia de Sistemas e Computadores of the Instituto Superior Técnico (both in Lisbon, Portugal). The data used are provided by the six other Portuguese institutions listed below:
- Phenotype data – Hospitais da Universidade de Coimbra (Coimbra), Centro de Cardiologia da Universidade de Lisboa, Hospital da Luz and Hospital de Sta. Cruz (all in Lisbon)
- Genotype data – Centro de Química Estrutural of the Instituto Superior Técnico of the Universidade Técnica de Lisboa and the Faculdade de Farmácia of the Universidade de Lisboa (both in Lisbon).
Link: http://kdbio.inesc-id.pt
Please contact:
Ana Teresa Freitas
KDBIO research group,
INESC-ID/IST Lisbon, Portugal
Tel: +351 213100394
E-mail:
