








by José García-Nieto and Enrique Alba
DNA microarrays have emerged as powerful tools in current genomic projects since they allow scientists to simultaneously analyse thousands of genes, providing important insights into the functioning of cells. Owing to the large volume of information that can be generated by a microarray experiment, the challenge of extracting the specific genes responsible for a given illness can only be solved by using automatic means. This challenge has driven our research group at the University of Málaga to design swarm intelligence approaches with the aim of performing accurate biological selection from gene expression datasets (AML-ALL leukemia, colon tumour, lung cancer, etc.).
A DNA microarray consists of a series of thousands of DNA molecules located in different positions within a matrix structure. These DNA molecules are mixed with cellular cDNA molecules during a hybridization process, after which, abundant sequences generate strong signals while rare sequences generate weak signals. Microarrays are normally used to compare gene expression intensity within a sample, and to look at differences in the expression of specific genes among samples. A sample is a test focussing on one disease or on healthy-unhealthy tissues. This is especially appropriate in cancer analysis, since it allows discrimination between tumour tissue and normal tissue. Several gene expression profiles obtained from tumours such as leukaemia, colon, and lung cancers are ready for research in computational biology, and we use them in our research.
The vast amount of data involved in a typical microarray experiment usually requires complex statistical analyses, with the goal of performing a supervised division of the dataset into correct classes. The key issue in this classification is to identify representative gene subsets that may be later used to predict class membership for new external samples. These subsets should be as small as possible in order to develop fast processes for the future class prediction done in an actual laboratory. The main difficulty in microarray classification versus classification of other datasets (found in other domains) is the availability of a relatively small number of samples in comparison to the huge number of genes in each sample (a typical microarray can have a few tens of tested sampled for several thousands of genes). In addition, expression data are highly redundant and noisy, and most genes are believed to be uninformative, as only a small proportion of genes may present distinct profiles for different classes of samples
In this context, machine learning techniques have been applied to handle large datasets, since they are capable of isolating useful information by rejecting redundancies. In practice, computational feature selection (gene selection, in biology) is often considered as a necessary pre-process step prior to analysing large datasets, in order to reduce the size of the dataset. Feature selection for gene expression analysis often uses supervised classification methods such as K-Nearest Neighbour (K-NN), Support Vector Machines (SVM), and Artificial Neural Networks (ANN) to discriminate a type of tumour. Nevertheless, optimal feature selection is a complex problem that has proved to be NP-hard, and therefore, efficient, automated and intelligent approaches are needed to tackle it.
Our research addresses all these issues. We use swarm intelligence algorithms in order to perform an efficient gene selection from large microarray datasets. Swarm intelligence approaches are computational procedures that model the collective behaviour of decentralized and self-organized systems found in nature (ant colonies, bee swarms, bird flocking, etc.) to solve an optimization or search problem. Using this approach, it is possible to reach optimal or quasi-optimal solutions to a given problem. Our goal is to minimise classification error whilst using the smallest possible set of genes to explain the results provided by a microarray. Swarm intelligence can result in savings of both time and resources in comparison to exhaustive and traditional search techniques. Essentially, our model consists of a particle swarm optimization (PSO) algorithm, in which a feature selection mechanism facilitates identification of small samples of informative genes among thousands of genes.
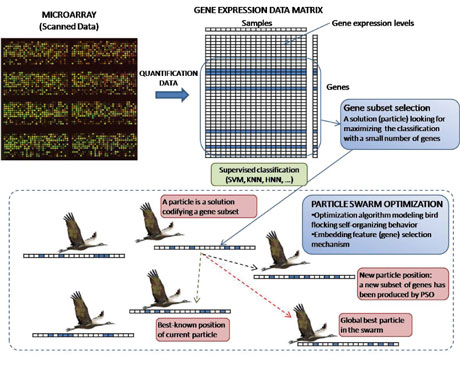
Figure 1: Complete process for optimal gene selection. Once the expression data are generated, our swarm intelligence algorithm obtains optimal subsets of representative genes that are offered to the human specialist as the genes responsible for the illness.
As shown in Figure 1, reported solutions by PSO (codifying gene subsets) are evaluated by means of their classification accuracy using SVM and cross-validation. The contribution of our swarm intelligence approach is notable, since it offers an improvement on existing state of the art algorithms in terms of computational effort and classification accuracy (see the links section). Furthermore, the gene ensembles found by this technique can be successfully interpreted in the light of independent existing results from biology, ie they are biologically (not just computationally) meaningful.
Link:
NEO Research Group: http://neo.lcc.uma.es
Please contact:
José García-Nieto
University of Málaga/SpaRCIM, Spain
Tel: +34 952133303
E-mail:
Enrique Alba
University of Málaga/SpaRCIM, Spain
Tel: +34 952132803
E-mail: