









by Jürgen Haas and Torsten Schwede
A linear sequence of amino acid letters or a three-dimensional arrangement of atoms in a polypeptide chain? Most biologists and bioinformaticians will have their preferred view when imagining a “protein”. Although these views represent two sides of the same coin, they are often difficult to reconcile. There are two main reasons for this: a lack of experimental structural information for the majority of proteins, and a different “culture” in handling data between the two communities, which results in a number of technical hurdles for somebody trying to bridge the gap between the two paradigms. Protein homology modelling resources establish a natural interface between sequence-based and structure-based approaches within the life science research community.
Natural proteins are exciting molecules which - in contrast to the regular helical DNA molecule - have characteristic, well-defined three-dimensional structures. It is the individual structure of a protein, with its intricate network of atomic interactions formed by the backbone and side chains atoms of the amino acids in a polypeptide chain, which allow it to perform highly specific functions in a living organism. These include mechanical force generated by motor proteins, enzymes degrading nutrients, antibodies recognizing foreign substances such as a pathogen, or olfactory receptors sensing the smell of perfume in one’s nose. But the most exciting property of these molecular machines is that they don’t need specific tools to be assembled: The linear sequence of amino acids alone contains sufficient information to define the three-dimensional structure when a protein is synthesized by a cell.
Mind the gap
Crucial to the understanding of molecular functions of proteins are insights gained from their three-dimensional structures. However, while thousands of new DNA sequences coding for proteins are sequenced every day, experimental elucidation of a protein structure is still an expensive and laborious process, typically taking several weeks. Not surprisingly, direct experimental structural information is, to date, only available for a small fraction of all proteins, and this gap is widening rapidly.
Whilst theoretically a nearly unlimited number of amino acid sequence combinations are possible, the number of different three-dimensional protein structures (“folds”) actually observed in nature is limited. This allows for homology (or comparative) modelling methods to build computational models for proteins (“targets”) based on evolutionary related proteins for which experimental structures are known (“templates”). Hence experimental structure determination efforts and homology modelling complement each other in the exploration of the protein structure universe.
The SWISS-MODEL Expert System
For an experimental scientist, being interested in obtaining a three-dimensional structural model of a protein to study a biological question should not require becoming an expert in molecular modelling or programming the necessary software tools. The aim of the SWISS-MODEL expert system is to provide a user-friendly Web-based system for protein structure homology modelling which is usable from any PC equipped with an Internet connection – without the need for installing complex software packages or huge databases. Sixteen years ago, SWISS-MODEL was the first fully automated protein modelling service on the Internet, aiming to make protein structure modelling easily accessible to life science research. Today SWISS-MODEL is one of the most widely used Web-based modelling services world wide. SWISS-MODEL hides the complexity of a stack of specialized modelling software, mirrors public databases of protein sequences and structures, automates procedures for data updates, and has tools for result visualization. Users interact with a set of partially or fully automated workflows in a personalized Web-based workspace. SWISS-MODEL is developed by the Computational Structural Biology Group at the Swiss Institute of Bioinformatics (SIB) and the Biozentrum of the University of Basel, Switzerland.
The Protein Model Portal
Diversity is essential to the success of any biological entity. However, there is reason to doubt that the same is true for the technical diversity observed among typical bioinformatics resources, especially in the field of molecular modelling. A simple question like “What is known about the three-dimensional structure of a given protein?” can easily end in an hour-long odyssey through the Internet for proteins which are not fully experimentally characterized. While all experimental data is collected centrally in the wwPDB – currently approximately 60,000 experimental structures are deposited in this database - computational structure models are generated by many specialized computational modelling groups spread out in the academic community. The heterogeneous user interfaces and formats used at different sites using various incompatible accession code systems are an additional challenge in accessing structure model information.
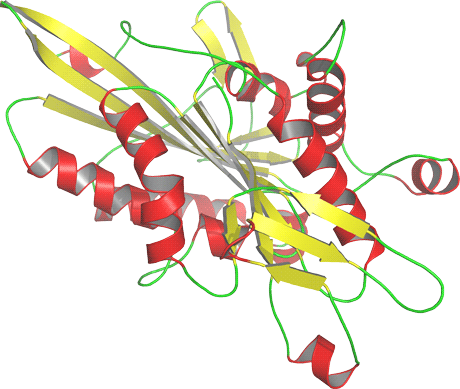
Figure 1: Three-dimensional homology model of the human Kinesin-like protein KIF3A, a protein involved in the microtubule-based translocation. The model was generated using the experimental structure of a related protein, the motor domain of the human Kinesin-like protein KIF3B sharing 66% sequence identity, as template.
In order to provide a single interface to access both experimental structures and computational models for a protein we have thus developed the Protein Model Portal (PMP) by federating the available distributed resources. PMP is part of the Nature – PSI Structural Biology Knowledgebase (PSI-SBKB) project, and is developed by the Computational Structural Biology Group at the Swiss Institute of Bioinformatics (SIB) and the Biozentrum of the University of Basel, Switzerland in collaboration with the PSI SGKB partner sites, specifically Rutgers, The State University of New Jersey. The PSI-SGKB informs about advances in structural biology and structural genomics in an integral manner, including not only newly determined three-dimensional structures, but also the latest protocols, novel materials and technologies. The current release of the portal (May 2010) consists of 12.7 million model structures provided by different partner resources for 3.4 million distinct protein sequences. PMP is available at http://www.proteinmodelportal.org and from the PSI Structural Biology Knowledgebase (http://kb.psi-structuralgenomics.org).
In order to integrate the meta-information on protein models available at the different partner sites – using heterogeneous data structures and incompatible naming conventions and accession code systems – we created a common independent reference system based on cryptographic md5 hashes for all currently known, naturally occurring amino acid sequences. This database is continuously updated while new sequences are being discovered, and allows us to dynamically federate information from different providers, without the need for the distributed resources to change their mode of operation. Using portal technologies, queries are transparently mapped to various database accession code systems, providing dynamic functional annotation for the target sequences.
For the first time it is now possible to query all participating federated structure resources - both experimental and computational models - simultaneously and compare the available structural information in a single interface. The dynamic mapping of the growing universe of all protein sequences even allows searching for features which were not implemented in the original data sources. Sometimes, the whole is greater than the sum of its parts.
Links:
http://www.proteinmodelportal.org
http://kb.psi-structuralgenomics.org
http://www.wwpdb.org
Please contact:
Jürgen Haas and Torsten Schwede
Biozentrum University of Basel
SIB Swiss Institute of Bioinformatics
Tel: +41 61 2671581
E-mail:
