









by Michal Dabrowski, Jakub Mieczkowski, Jakub Lenart and Bozena Kaminska
Understanding how multiple genes change expression in a highly ordered and specific manner in healthy and diseased brains may lead to new insights into brain dysfunction and identification of promising therapeutic targets, for example key signalling molecules and transcriptional regulators. To this end, we have been performing integrated analyses of high-throughput datasets, genomic sequence-derived data and functional annotations.
The computational work of our group (Laboratory of Transcription Regulation, The Nencki I
stitute) focuses on developing tools that permit a systemic view of global transcriptional/epigenetic changes and applying these tools to the data from experimental models studied in our laboratory, in particular: animal models of stroke, brain tumours and in vitro experimental models of brain tumour-host interactions. In this way computational results can guide experimental effort, which in turn verifies the computational predictions.
Computational/bioinformatics activities of our group focus on the following areas:
- building a comprehensive database of predicted or experimentally identified cis-regulatory regions, transcription factor binding sites (TFBS) and chromatin modifications in vertebrates (human, mouse, rat)
- functional interpretation of global gene expression or other high-throughput data in the context of databases of functional annotations (Gene Ontology) or biological pathways (KEGG)
- identification of transcriptionally co-regulated genes from global gene expression, genomic sequence-derived and chromatin modification data.
We have previously established a local Transcription regulatory regions and motifs (TRAM) database; of putative transcription regulatory regions (AVID-VISTA conserved non-coding regions) and TFBS motif instances (Genomatix) for all pairs of corresponding (orthologous) genes in human and rat. This database is now being updated, extended, and prepared for future automated actualization and public release, as a part of the Nencki Regulatory Genomics Portal (NRGP) project, started in early 2010 and scheduled for four years. The data layer of the NRGP will incorporate additional vertebrate species; and alternative sources of putative regulatory regions; both computationally predicted (BlastZNet, cisRED promoters) and established experimentally (Enhancer VISTA, Ensembl funcgen). These regulatory regions will be scored with each of the three major TFBS motif libraries, both commercial (Transfac, Genomatix) and public (JASPAR). We will implement an innovative way of mapping regulatory regions to the genes, taking into account positions of insulators. The data layer of NRGP is illustrated in Figure 1A.
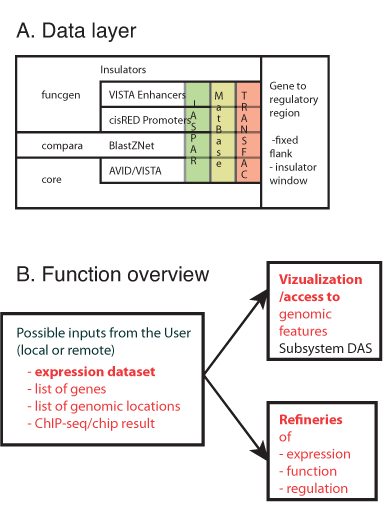
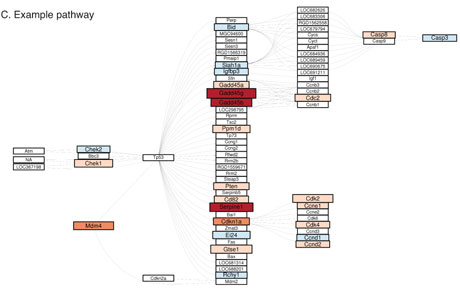
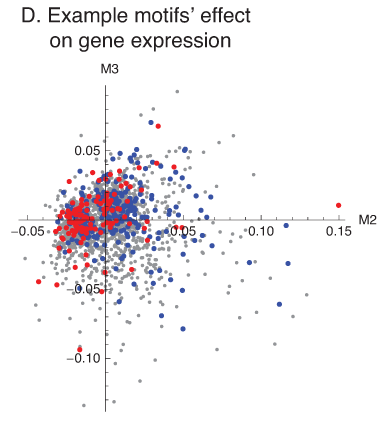
Figure 1: Functionality of the future Nencki Regulatory Genomics Portal.
Nencki Regulatory Genomics Portal will be an internet service providing access to the its data layer and tools for analysis of user-supplied gene expression data, aiming at identification of functions and/or cis-regulatory mechanisms common to many genes. The service will be available by web browser for the users want to analyze the data with the help of provided algorithms, and by a database client for the ones who need direct access to the stored information. DAS technology will be used to make our data layer accessible from the Ensembl genome browser.
Architecture of NRGP, based on MVC (Model-View-Control) model, will consist of the following layers:
- Data layer – responsible for all data storage and actualization corresponding to changes made in its sources like Ensembl or motif libraries
- Application layer – divided into the front-end responsible for implementation of main algorithms associated with the refinement of gene expression, regulation and function; and the back-end – providing services related to the actualization, like searching for the TFBS motifs
- Presentation layer – responsible for GUI (Graphical User Interface), adapted to run in web browser.
In order to achieve high level of quality of supported analysis NRGP will integrate worldwide known and used components like Mathematica, R statistical programming language, and commercial libraries. Moreover, the team developing the service consists of experts in scientific areas like: biology of gene expression, and informatics of software engineering, and mathematics of machine learning and data mining.
NRGP will provide the following functionalities (see Figure 1B):
- Refinement of expression – starting from an expression dataset uploaded by the user, this module permits identification and visualization of common patterns of gene expression, with standard techniques of analysis microarray gene expression data (analysis of variance, several clustering algorithms, SVD/PCA). In a recent paper, linked below, we show that probe set filtering increased correlations between microarray and qRT-PCR results in two types of studies: detection of differential expression computed by p-values of t-test and estimation of fold change between analyzed groups.
- Refinement of function – permits identification of functional annotations statistically associated with a set of genes with a particular pattern of expression, with standard techniques (Fisher Exact Test, Wilcoxon signed rank test). We are currently working on a novel analysis of changed signal (ACS) algorithm for identification of signalling pathways affected by changes in gene expression, which takes into account established topology of signalling pathways, is threshold-free, and does not require the assumption of independence between genes. Figure 1C shows visualization of changes in expression of the genes in a particular signalling pathway
- Refinement of regulation – aims at identification of TFBS motifs and/or chromatin modifications associated with a particular pattern of gene expression, by finding motifs over-represented in putative regulatory regions (Fisher Exact Test), followed by linear or logistic regression to study the effect of motif multiplicity on the given pattern of expression. We previously reported analysis of cis-regulation in subspaces of conserved eigensystems (bi-orthogonal components, also called SVD modes). In a recent paper, linked below, we demonstrate how bi-orthogonality of gene expression data can emerge as a result of biological processes occurring in different cell types, with signal passing between them. In collaboration with Dr. Norbert Dojer (Institute of Informatics, University of Warsaw) we used our method, in the framework of Bayesian Networks learning, to dissect transcriptional regulation in brain following stroke and seizures (Figure 1D). Moreover, we show that effects of motif multiplicity on gene expression analyzed in subspace follow the predictions of the linear response model of gene regulation.
In our research we collaborate with the Computational Biology Group led by Prof. Jerzy Tiuryn (Institute of Bioinformatics, Uniwersity of Warsaw) and with the Linneus Center for Bioinformatics (Uppsala, Sweden) directed by Prof. Jan Komorowski.
Links:
http://www.nencki.gov.pl/pl/struktura/biologia_komorki/lab_02.html
http://www.biomedcentral.com/1471-2105/7/367
http://www.biomedcentral.com/1471-2105/11/104
Please contact:
Michal Dabrowski
The Nencki Institute, Poland
Tel: 4822 58 92 232
E-mail:
