









by Witold R. Rudnicki and Łukasz Ligowski
Graphic processors are used to improve the efficiency of a computational process in molecular biology in a project carried out by our team at the Interdisciplinary Centre for Mathematical and Computational Biology of the University of Warsaw, in cooperation with Professor Bertil Schmidt’s team from the School of Computer Engineering of the Nanyang Technological University in Singapore. The project aims to increase the efficiency of one of the most important algorithms in bioinformatics, the Smith Waterman algorithm.
The Smith Waterman (SW) algorithm, an algorithm for performing local sequence alignment, is used to establish similarity between biological macromolecules, such as proteins and nucleic acids. This is an exact algorithm giving the best possible result, but is much slower than BLAST (Basic Local Alignment Search Tool) - an alternative heuristic algorithm giving less precise answers but in a fraction of time. Availability of the fast version of SW algorithm with efficiency comparable to that of BLAST would enable researchers to take advantage of the greater sensitivity of the SW algorithm in the tasks currently performed with heuristic algorithms. This may be particularly important, for example, for gene assembly from short fragments in the new generation sequencing experiments or high sensitivity search for homologous proteins.
The evolution of graphics processing units (GPU) has resulted in transformation from chips specialised in the limited number of operations required for graphics, to massively parallel processors capable of delivering trillions of floating point operations per second (teraflops). The GPU’s very high computational power is best utilised when and all threads perform identical tasks requiring many operations on the relatively small amount of data, which can be stored on on chip (in registers and a small pool of the very fast shared memory) The best practice in GPU programming is therefore to use data parallelism to formulate the problem, load the data to registers and shared memory, and perform all computations utilising registers and shared memory.
GPU has the potential to be particularly effective for applications such as scanning of large databases of sequences of biological macromolecules, because GPU works best for data-parallel algorithms, with a small but computationally intensive kernel.
Optimisation of the SW algorithm on a central processing unit (CPU) is based on vectorization and improving efficiency of the alignment of a single pair of sequences using some tricks, the efficacy of which is data-dependent. In particular these optimisations work poorly on short sequences, as well as on sets of sequences which are similar to one other. A GPU version of the algorithm, in contrast, performs a more efficient data base scan by performing thousands of individual scans in parallel. No data-dependent optimisations are employed, hence the performance of the algorithm is stable both for similar and short sequences.
Our algorithm scans several thousand sequences in parallel. The smallest chunk of sequences which are processed together comprises 256 sequences. These are processed on a single multiprocessor. The chips of current high-end GPUs contain at least 16 identical multiprocessors, therefore a GPU performs alignment of at least 4096 sequences in parallel.
Protein sequences are represented as a string of 20 characters corresponding to 20 types of amino acid. Homology between proteins is inferred from the similarity of their sequences - if sequence similarity is higher than expected by chance then the proteins are homologous. The alignments can contain gaps – these represent mutations, which correspond to deletions or insertions in the proteins. The score of the alignment is the sum of the similarity scores for amino acid pairs along alignment after subtracting the penalties for introducing gaps. The similarity score for a pair of amino acids is proportional to the logarithm of probability of mutation between these amino acids in proteins.
The Smith Waterman algorithm finds best local alignment by identifying all possible alignments. From among these it selects the optimal alignment. This is achieved using a dynamic programming approach. First a rectangular matrix is built, then each cell with index [i,j] is filled with a similarity score for the i-th residue of the query sequence and j-th residue of the target sequence. Then, starting from the upper left corner corresponding to the start of both sequences, one determines the best possible alignment ending in each cell. An appropriate score is then assigned to the cell. To process the cell one needs to know information on score and auxiliary variable in three adjacent cells - upper, left and upper-left.
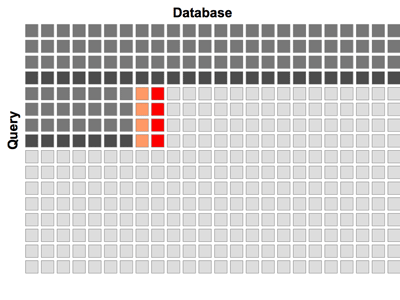
Figure 1: Processing of the dynamic programming matrix in horizontal bands. Cells, which are already processed, active cells and cells waiting for processing are displayed in dark gray, red and light gray, respectively. Data in orange cells to the left of active cells is stored in shared memory. Cells containing data accessed on beginning of the loop are highlighted in deep dark gray.
In our algorithm the SW matrix is processed in 12-cell high bands, as shown in Figure 1. Each band is processed column by column. The global memory access is only required twice, once at the entrance to the main loop and once at the exit. The score and auxiliary variable from the preceding column are stored in the shared memory. The algorithm achieves an approximately 100-fold increase in speed compared withthe single core of CPU, and its overall efficiency is on par with BLAST - the heuristic algorithm executed on CPU.
The sw-gpu algorithm is used as an engine of our similarity search server accessible at http://bioinfo.icm.edu.pl/services/sw-gpu/
We are currently working on using SW algorithm in an iterative way, analogous to the PSI-BLAST extension of the BLAST algorithm. In this approach the results of the search are used to develop an improved position-specific similarity function, which is then used for a refined search. The procedure is repeated several times, until no new sequences are found. Another application field is assembling of the DNA/RNA sequences from short fragments which are generated in the new sequencing methods (next generation sequencing).
Link: http://bioinfo.icm.edu.pl/services/sw-gpu/
Please contact:
Witold R. Rudnicki,
University of Warsaw, Poland
Tel: +48 22 5540 817
E-mail:
