








by Claudia Angelini, Alfredo Ciccodicola, Valerio Costa and Italia De Feis
The recent introduction of Next-Generation Sequencing (NGS) platforms, able to simultaneously sequence hundreds of thousands of DNA fragments, has dramatically changed the landscape of genetics and genomic studies. In particular, RNA-Seq data provides interesting challenges both from the laboratory and the computational perspectives.
Gene transcription represents a key step in the biology of living organisms. Several recent studies have shown that, at least in eukaryotes, virtually the entire length of non-repeat regions of a genome is transcribed. The discovery of the pervasive nature of eukaryotic transcription and its unexpected level of complexity - particularly in humans – is helping to shed new light on the molecular mechanisms underlying inherited disorders, both mendelian and multifactorial, in humans.
Prior to 2004, hybridization and tag-based technologies, such as microarray and Serial/Cap Analysis of Gene Expression, offered researchers intriguing insights into human genetics. Microarray techniques, however, suffered from background cross-hybridization issues and a narrow detection range, whilst tag-based approaches required laborious time- and cost-effective steps for the cloning of fragments prior to sequencing. Hence, the recent introduction of massively parallel sequencing on NGS platforms has completely revolutionized molecular biology.
RNA-Seq is probably one of the most complex of the various “Seq” protocols developed so far. Quantifying gene expression levels within a sample or detecting differential expressions among samples, with the possibility of simultaneously analysing alternative splicing, allele-specific expression, RNA editing, fusion transcripts and expressed single nucleotide polymorphisms, is crucial in order to study human disease-related traits.
To handle this novel sequencing technology, molecular biology expertise must be combined with a strong multidisciplinary background. In addition, since the output of an RNA-Seq experiment consists of a huge number of short sequence reads - up to one billion per sequencing run - together with their base-call quality values, terabytes of storage and at least a cluster of computers are required to manage the computational bottleneck.
Recently, the Institute of Genetics and Biophysics (IGB) and the Istituto per le Applicazioni del Calcolo (IAC), both located in the Biotechnological Campus of the Italian National Research Council in Naples, have started a close collaboration on RNA-Seq data which aims to fill the gap between data acquisition and statistical analysis. In 2009 IGB, a former participant in the Human Genome project, acquired the SOLiD system 3.0, one of the first and most innovative platforms for massively parallel sequencing installed in Italy. IAC has great experience in developing statistical and computational methods in bioinformatics and is equipped with two powerful clusters of workstations capable of handling massive computational tasks.
The collaboration started with two pilot whole-transcriptome studies on human cells via massively parallel sequencing aimed at providing a better molecular picture of the biological system under study and at setting up an efficient computational open-source pipeline to downstream data analysis.
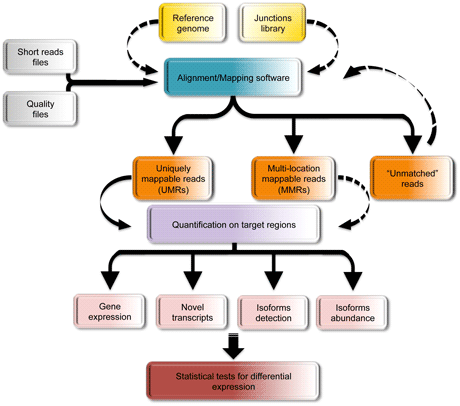 Figure 1: An example of the computational pipeline for the analysis of RNA-Seq data.
Figure 1: An example of the computational pipeline for the analysis of RNA-Seq data.
The computational effort focuses on the use of efficient software, the implementation of novel algorithms and the development of innovative statistical techniques for the following tasks:
a) alignment of the short reads to the corresponding reference genome
b) quantifying gene expressions
c) procedures of normalization to compare samples obtained in different runs and assessment of the quality of the experiments
d) identification of novel transcribed regions and refinement of previously annotated ones
e) identification of alternative spliced isoforms and assessment of their abundance;
f) detection of differential genes/isoform expression under two or more experimental conditions
g) implementation of user-friendly interfaces for data visualization and analysis.
Each of these tasks requires the integration of currently available tools with the development of new methodologies and computational tools. Despite the unprecedented level of sensitivity and the large amount of data available to provide a better understanding of the human transcriptional landscape , the useful genetic information generated in a single experiment clearly represents only “the tip of the iceberg”. Much more research will be needed to complete the picture.
For steps a) and f), we are integrating some open source software into our pipeline. These are well-consolidated phases and there are several methods available in the literature. Points b) – e) are far more difficult as statistical methodologies are still lacking. The mathematical translation of the concept of “gene expression” and its modelling needs to be reassessed since we are now faced with discrete variables; we are now applying innovative methods.
The results we obtain from the computational analysis of the two pilot projects will be validated by quantitative real-time polymerase chain reaction (PCR) and, where deemed crucial for the analysis, the related protein products will be assessed by Western Blot. Biological validation will provide fundamental feed-back for optimizing the parameters of the computational analysis.
Please contact:
Claudia Angelini or Italia De Feis
IAC-CNR, Italy
E-mail:
Alfredo Ciccodicola or Valerio Costa
IGB-CNR, Italy
E-mail: