







by Eleftherios Christofi (The Cyprus Institute) Vagelis Harmandaris (The Cyprus Institute, University of Crete and FORTH-IACM)
By enhancing multi-scale molecular simulations with deep learning, AI is enabling researchers to bridge modelling scales that were once out of reach. This synergy opens new possibilities for understanding complex materials and accelerating scientific discovery.
Understanding complex macromolecular systems requires computational tools that can capture their behaviour on a wide range of spatio-temporal (length and time) scales. Classical atomistic simulations provide a valuable tool for exploring the structure and dynamic evolution of polymeric systems at the molecular scale, but they face fundamental limitations on the accessible length and time scales, mainly due to the long relaxation times required for equilibration. To overcome such limitations, a number of different approaches have been employed to model systems across different levels of detail. Hierarchical particle-based computational modelling of macromolecular materials across various scales is based on the systematic exchange of information between different (e.g. microscopic and mesoscopic) representations of atoms and molecules to capture material properties for a wide range of spatial and temporal scales. By considering models at different scales simultaneously, one hopes to develop an approach that shares the efficiency of the coarser models, as well as the accuracy of the microscopic (finer) ones.
Therefore, such approaches are nowadays applied to a wide range of polymer-based molecular systems in various fields such as physics, chemistry, biology, materials science, and engineering. A common characteristic of all the above systems is that they are characterised by a very broad range of spatio-temporal scales. Hence, to predict their properties and obtain a fundamental understanding of their behaviour, simulation methods over different scales are necessary.
Artificial intelligence (AI), and deep learning in particular, is now offering new opportunities to advance these multi-scale approaches. Neural networks can learn structural patterns directly from simulation data, allowing them to represent complex molecular information in ways that traditional rules or functional forms cannot easily achieve. This makes AI a valuable complement to classical modelling, especially in situations where information is lost during coarse-graining or when atomistic simulations become too costly.
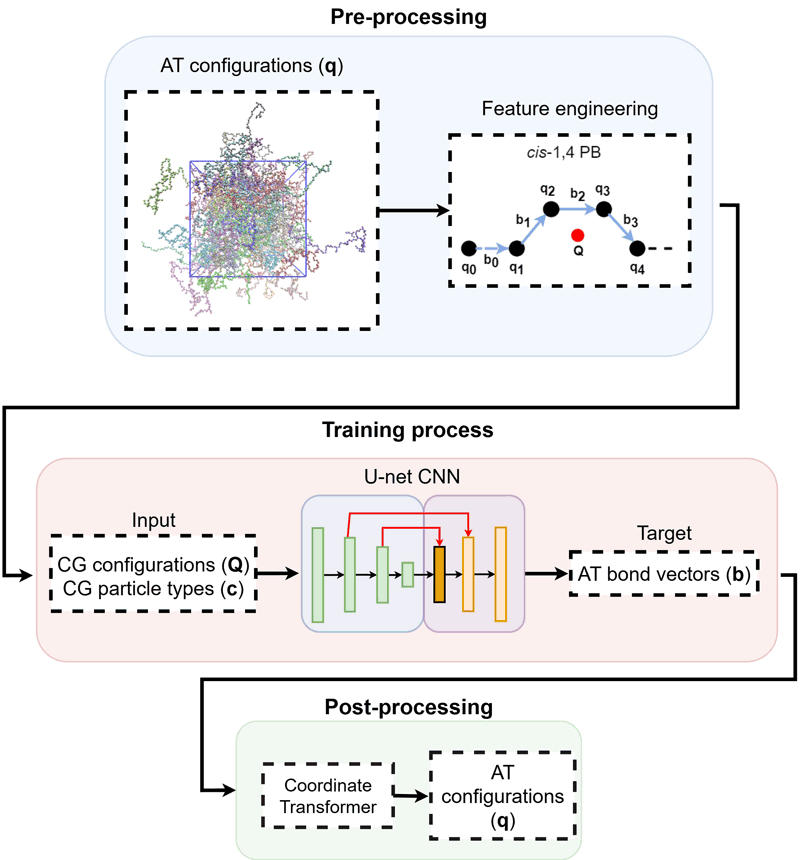
Figure 1: Schematic representation of the systematic back-mapping of the coarse-grained multi-component molecular system via deep learning models.
An important direction involves moving upwards in resolution. To expand the range of accessible scales via atomistic simulations, coarse-grained (CG) models that reduce the dimensionality of the physical system under study have been developed. In systematic bottom-up CG models, groups of atoms are lumped together into particles that are typically denoted as “superatoms” or beads. Currently, such approaches are applied to a multitude of molecular systems, such as proteins and synthetic polymers. However, when a molecular system is simplified to a coarser representation, much of its atomistic detail is removed. Therefore, in order to obtain information on properties that depend on the microscopic structure, an atomic-level resolution is necessary. Restoring this missing information is a difficult task, yet it is essential when fine-scale accuracy is needed. Recently, we have developed a general DL-based approach for back-mapping (re-introducing atomic detail) in CG multi-component macromolecular systems, based on convolutional neural networks (CNN) that are trained directly on atomistic descriptors (see Figure 1). The new DL model is capable of creating representative well-equilibrated atomistic configurations in three-dimensional Cartesian space of multi-component polymeric materials of high molecular weight [1, L1] (see Figure 2). To improve the accuracy of the deep learning model, the loss function of the neural network is augmented with several penalizing terms based on prior knowledge of the physical properties of the molecular systems under study, such as bond lengths, bond angles, and dihedral angles. In addition, the proposed approach avoids tedious and labor-intensive bookkeeping of molecular details during the reconstruction by separating configurational information from molecular topology and force fields.
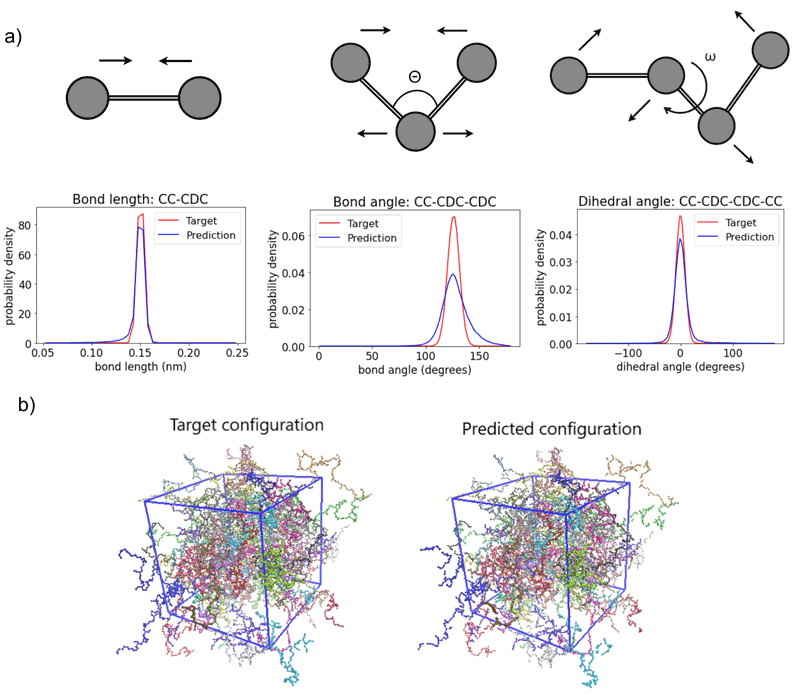
Figure 2 (a) Comparisons of bond lengths, bond angles, and dihedral angles among target atomistic configurations and predictions obtained via deep learning models. (b) Target and predicted (through deep learning models) snapshots of a bulk polymeric system generated through deep learning models.
Multi-scale modelling can also be used to make predictions that move downwards in resolution, from detailed molecular structure towards macroscopic behaviour. This is particularly relevant for polymer nanocomposites, where local structural heterogeneities and polymer–filler interactions strongly influence the overall mechanical response. Atomistic simulations are capable of resolving per-atom stress and strain fields, yet the computational cost of generating these fields for multiple systems and load cases is substantial. To address this challenge, a data-driven model was developed that predicts these mechanical fields directly from structural descriptors extracted from atomistic trajectories [2, L2]. The model learns to reproduce the spatially varying mechanical response across the matrix, interphase, and nanoparticle regions, allowing researchers to examine how filler content, polymer configuration, and interfacial structure affect the emergent mechanical properties without performing new large-scale simulations for each case.
Together, these directions demonstrate how AI can support multi-scale modelling by improving the flow of information between different levels of resolution. Deep learning assists in reconstructing the fine-scale atomistic structure when needed and accelerates the prediction of macroscopic properties originating from microscopic features. In this way, AI driven models help balance efficiency and accuracy, allowing researchers to explore large systems while still retaining access to detailed molecular information. Building on these methodologies, future work will focus on developing models to explore the chemical design space, which can be used to support the in-silico design of materials with targeted macroscopic properties.
As AI-powered modelling continues to mature, its integration with multi-scale approaches is expected to deepen, opening new possibilities for the design and understanding of advanced molecular materials. The combination of hierarchical modelling and machine learning provides an effective route towards bridging previously inaccessible scales and offers a promising foundation for future advances in macromolecular simulation.
Our research team is an interdisciplinary team composed of chemical engineers and applied mathematicians. We have long-standing experience in model reduction methods, mathematical and computational modelling of complex systems, and scientific machine learning.
Three different institutes are involved in the research project: the University of Crete (Greece), the Institute of Applied and Computational Mathematics – FORTH (Greece) and the Cyprus Institute (Cyprus). We also have a long-term’ active collaboration with research institutes around the world.
Links:
[L1] https://github.com/SimEA-ERA/CNN-BackMap-CG
[L2] https://github.com/SimEA-ERA/DeepNANOMEC
References:
[1] E. Christofi et al., “Deep convolutional neural networks for generating atomistic configurations of multi-component macromolecules from coarse-grained models”, J. Chem. Phys., 2022, 157, 184903. https://doi.org/10.1063/5.0110322
[2] E. Christofi et al., “Physics-informed deep learning model for predicting the heterogeneous mechanical properties of polymeric nanostructured materials”, SSRN, 2025. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5372731
Please contact:
Vagelis Harmandaris, Eleftherios Christofi
The Cyprus Institute, Cyprus, University of Crete, Greece, FORTH-IACM, Greece