







by Adrián Segura Ortiz, José García-Nieto and Ismael Navas Delgado (ITIS Software, University of Málaga)
The resulting consensus networks are more robust and interpretable, enabling deeper insights into complex disease mechanisms.
Understanding how genes interact with each other is essential for explaining how cells function and why diseases develop. Advances in molecular biology now enable us to measure thousands of genes simultaneously. However, transforming these measurements into accurate maps of regulatory interactions remains a significant challenge. Different computational algorithms tend to disagree, and many overlook well-established biological principles, resulting in models that are mathematically correct but biologically implausible.
Responding to this need, researchers at the University of Málaga (Spain), within the Khaos Research Group [L1] and the ITIS Software Institute [L2], have developed a framework for context-guided consensus inference of gene regulatory networks (GRNs). Instead of relying on a single method, the project combines multiple existing techniques and aligns them with biological knowledge. Individual algorithms are treated as complementary “opinions”, and consensus networks are produced through evolutionary optimisation guided by biological realism rather than purely mathematical scores (Figure 1).
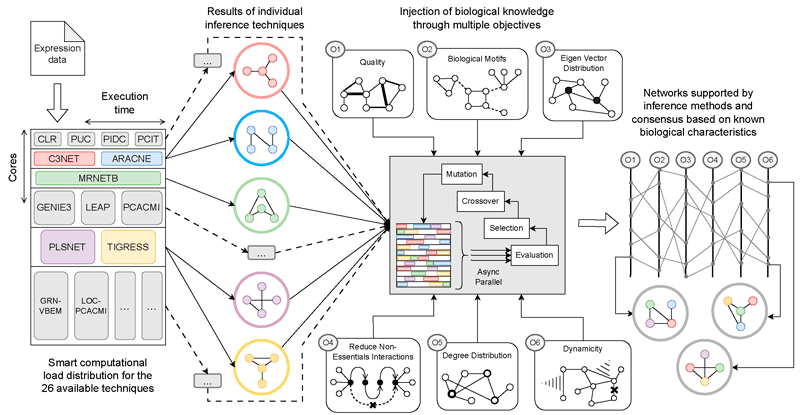
Figure 1. Overview of the context-guided consensus proposal for inferring gene regulatory networks. Multiple inference techniques are executed in parallel, each producing a different candidate network. These networks are evaluated and combined through an evolutionary process informed by biological knowledge, including regulatory patterns, interaction quality, and realistic network structures. The resulting consensus networks are both supported by the computational evidence and constrained by known biological characteristics.
The initiative began in 2022 as part of a doctoral thesis and progressed through a sequence of increasingly biologically guided tools:
- GENECI [1], the first release, demonstrated that evolutionary search could combine predictions from multiple GRN inference techniques. Using a simple genetic algorithm, it produced more stable networks than individual methods alone, showing that consensus could be treated as an optimisation problem rather than a voting procedure.
- Memetic Inference extended this idea by incorporating a small number of known gene–gene interactions into the optimisation process. A local search component helped refine the networks towards these known links, improving accuracy without overfitting to prior knowledge.
- MO-GENECI [2] then shifted from a single optimisation goal to a multi-objective interpretation, incorporating structural and functional properties often observed in real regulatory systems, such as characteristic topologies and regulatory motifs. This allowed the algorithm to preserve agreement among methods while also favouring biologically plausible organisation.
- PBEvoGen brought domain experts into the loop. Rather than merely interpreting results, specialists could steer the optimisation toward regions of biological interest, transforming consensus inference into an interactive process guided by expert knowledge.
- Finally, BIO-INSIGHT [3] generalised this approach into a many-perspective strategy, simultaneously evaluating regulatory networks from several biological viewpoints (including expected interaction densities, dynamic behaviour, motif enrichment, and reduction of spurious links) and scaling efficiently to large datasets through distributed computation.
These tools are not isolated prototypes. They have been unified in GENECI, an open-source Python package [L3] that integrates the orchestrator of more than 25 GRN inference techniques with the full evolutionary framework. The package is freely available to the community [L4] and has already been downloaded over 16,000 times, demonstrating interest from research groups working in gene regulation, computational biology, and clinical data mining.
This progression has been driven by a practical need: in biomedical contexts, biological relevance takes precedence over numerical fit. A network may achieve excellent statistical performance yet fail to represent meaningful regulatory behaviour. For example, some algorithms prioritise noise reduction and lose regulatory motifs, while others perform well in yeast but fail in human tissue. By favouring biologically coherent interactions, the framework reduces false positives and improves interpretability. Its aim is to generate credible hypotheses that scientists and clinicians can meaningfully investigate.
The framework has been validated using both simulated benchmarks and real-world clinical data. Collaborators provided transcriptomic datasets from melanoma, fibromyalgia, and myalgic encephalomyelitis. In these studies, the algorithms identified regulatory patterns associated with inflammation and immune dysregulation. While not designed for diagnosis, the results provide testable biological leads, supporting research into conditions where reliable biomarkers are still lacking.
Collaboration has been essential. A research stay at the University of Lille (France) contributed expertise in evolutionary computation, while clinical partners at the University of Valencia (Spain) provided access to medical data and biological interpretation. This cross-disciplinary model reinforces the project’s commitment to reproducibility: all methods are openly released, documented, and designed to be extended by the scientific community.
Looking ahead, the team plans to adapt consensus inference to single-cell transcriptomics, enabling analysis of regulatory variability between individual cells. They are also developing interactive tools that allow experts to guide optimization in real-time. Together with opportunities for cooperation within the ERCIM community in regulatory modelling, explainable AI, and biomedical graph mining, these developments highlight a broader ambition: to bridge computational innovation with biological insight and deepen our understanding of the regulatory mechanisms that govern life.
Links:
[L1] https://khaos.uma.es/
[L2] https://itis.uma.es/
[L3] https://pypi.org/project/geneci/
[L4] https://github.com/AdrianSeguraOrtiz/GENECI
References:
[1] A. Segura-Ortiz, et al., “GENECI: A novel evolutionary machine learning consensus-based approach for the inference of gene regulatory networks,” Comput. Biol. Med., vol. 155, Art. no. 106653, 2023.
[2] A. Segura-Ortiz, et al., “Multi-objective context-guided consensus of a massive array of techniques for the inference of gene regulatory networks,” Comput. Biol. Med., vol. 179, Art. no. 108850, 2024.
[3] A. Segura-Ortiz, et al., “Multifaceted evolution focused on maximal exploitation of domain knowledge for the consensus inference of gene regulatory networks,” Comput. Biol. Med., vol. 196, Art. no. 110632, 2025.
Please contact:
Adrián Segura Ortiz
ITIS Software, University of Málaga, Spain
José Francisco Aldana Montes,
ITIS Software, University of Málaga, Spain