









by George Potamias (FORTH)
Within the framework of the Greek eMoDiA (electronic PharmacoGenomics Assistant) project, we developed an electronic pharmacogenomics assistance platform using machine learning techniques.
Pharmacogenomics (PGx) has revolutionised drug therapy and unearthed hundreds of associations between genes and drug response, with genome wide association studies (GWAS) and next generation sequencing (NGS) approaches to boost the engaged association discovery process [1]. In order to serve the needs of the different PGx communities – from biomedical researchers to clinical decision-makers and therapy planners – we designed and implemented an electronic PGx Assistance (ePGA) platform [L1], [2,3]. The whole endeavour was conducted in the context of the eMoDiA (Greek funded) project [L2].
ePGA offers two basic services: (i) explore – a browser-based service to search through established PGx associations and their accompanying metabolic phenotypes (i.e., extensive, intermediate, poor and ultra-rapid); (ii) translate (see Figure 1(a) – an algorithmic process that infers PGx metabolic phenotypes (referred also as a metaboliser statuses) from individual genotype profiles. In the heart of the translation process are the haplotype tables that define PGx haplotypes with reference to gene-variants. PharmGKB (a state-of-the-art PGx knowledge base, www.pharmgkb.org), provides haplotype tables for a total of 69 pharmacogenes (genes for which respective haplotypes are related with metabolising response profiles) that engage a total of 786 variants.
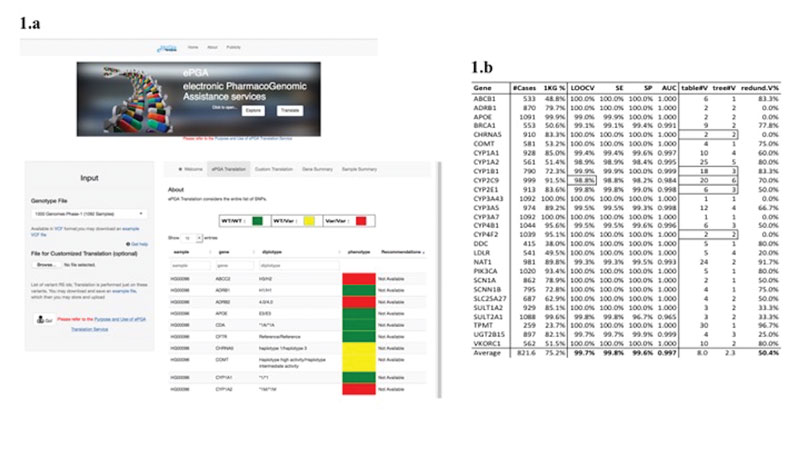
Figure 1: (a) The ePGA translation service; and (b) Results of PGx decision-tree prediction models (using just the variants in haplotype tables).
The ePGA translation process was applied on the phase-I 1000 Genomes (1kG) project samples (1092 in total, covering fourteen populations, www.1000genomes.org). For each gene, a sample is assigned to a PGx phenotype status, i.e., the class target variable, which may take one of the following three (phenotype status) values: “RR” (reference/wild-type), “RV” (reference-variant), or “VV” (variant-variant). The variants (rsIDs) represent the descriptor features that take values over all the possible allele combinations, e.g., the variant “rs12475068: C>G” may take the values CC, CG (or GC) and GG. So, we have at our disposal a genotype matrix with rows the variants, columns the samples, and cell values the genotypes of the respective samples. The target is to form patterns of variants that could model, and in a way explain the assignment (by the translation process) of phenotype class to the samples – a classical machine-learning problem, which we tackle with the utilisation of decision-tree induction techniques. We restricted our experiments to 28 genes for which at least 20% of the samples are assigned to one phenotypic class. For each gene the respective genotype data (restricted to the rsIDs present in the original gene haplotype table) were used as input to a decision-tree induction algorithm (the Weka J48 tool was utilised). For each gene a respective decision-tree was induced. The results are presented in Figure 1(b).
The high performance figures, LOOCV: 99.7%, SEnsitivity: 99.8%, SPecificity: 99.6% and ROC/AUC: 0.997 are indicative for the validity of the whole approach. The highly predictive results are achieved with a reduced set of variants – the percentage of common variants is 49.6% resulting in a 50.4% reduction in the number of utilised variants (column “redund.V%”). So, we can safely state that: using a representative set of genotyped samples we are able to identify a reduced number of “critical” and “informative” variants, and form respective predictive models (PGx-decision-trees) that are able to accurately infer the PGx phenotype phenotypic metaboliser status of the sample cases. We also applied the same approach on genotype profiles that include the whole set of variants present in the region of the respective genes. The high performance figures, LOOCV: 99.6%, SE: 96.1%, SP: 99.5% and AUC: 0.996, achieved with a common variants’ percentage of 60%, also confirm the validity of our decision-tree induction approach.
Our on-going and future R&D work in the field includes: (a) investigation of the ways to form new haplotype tables utilising the set of variants present in the respective PGx decision-trees, and (b) experimentation with more sets of population representative genotyped samples in order to achieve a more extended and deeper validation of the approach.
Links:
[L1] http://www.epga.gr
[L2] http://www.emodia.gr
References:
[1] G. Potamias K.Lakiotaki, T. Katsila, et al.: “Deciphering next-generation pharmacogenomics: an information technology perspective”, Open Biol. 4(7), 2014, doi: 10.1098/rsob.140071.
[2] K. Lakiotaki, G. Patrinos, G. Potamias: “Information Technology meets Pharmacogenomics: Design Specifications of an Integrated Personalized Pharmacogenomics Information System”, IEEE-EMBS Int. Conf. Biomed. Heal. Informatics, pp. 13–16, 2014.
[3] K. Lakiotaki, et al.: “ePGA: a web- based Information System for Translational Pharmacogenomics,” PLOS One 11(9):e0162801. doi:10.1371/journal.pone.0162801, 2016.
Please contact:
George Potamias, ICS-FORTH, Greece

