









by Torsten Schaub and Anne Siegel
We use expressive and highly efficient tools from the area of Knowledge Representation for dealing with contradictions occurring when confronting observations in large-scale (omic) datasets with information carried by regulatory networks.
The availability of high-throughput methods in molecular biology has led to a tremendous increase of measurable data along with resulting knowledge repositories, gathered on the web usually within biological networks. However, both measurements and biological networks are prone to considerable incompleteness, heterogeneity, and mutual inconsistency, making it difficult to draw biologically meaningful conclusions in an automated way.
Further probabilistic and heuristic methods exploit disjunctive causal rules to derive regulatory networks from high-throughput -static- experimental data. For instance, disjunctive causal rules on influence graphs were originally introduced in random dynamical frameworks to study global properties of large-scale networks, using a probabilistic approach. These were demonstrated mainly on the transcriptional network of yeast. However, these methods are mostly data driven, and they lack the ability to perform corrections in a fast and global way. In contrast, efficient model-driven approaches based on model checkers - such as multi-valuated logical formalisms - are available to confront networks and measured data. These however, make use of time-series observations and can only be applied to small-scale parametered systems, since they need to consider the full dynamics of the system.
We have proposed an intermediate approach to perform diagnosis on large-scale static datasets. We use a Sign Consistency Model (SCM), imposing a collection of constraints on experimental measurements together with information on cellular regulations between network components.
The main advantage of SCM lies in its global approach to confronting networks and data, since the model allows the propagation of static information along the network and localization of contradictions between distant nodes. In contrast to available probabilistic methods, this model is particularly well-suited for dealing with qualitative knowledge (for instance, reactions lacking kinetic details) as well as incomplete and noisy data. Indeed, SCM is based on influence (or signed interaction) graphs, a common representation for a wide range of dynamical systems, lacking or abstracted from detailed quantitative descriptions.
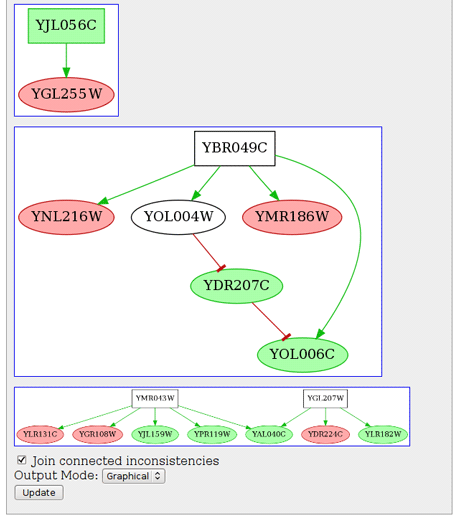
Figure 1: A graphical representation of identified inconsistencies in an Escherichia coli network.
By combining SCM with efficient Boolean constraints solvers, we address the problem of detecting, explaining, and repairing contradictions (called inconsistencies) in large-scale biological networks and datasets by introducing a declarative and highly efficient approach based on Answer Set Programming [1]. Moreover, our approach enables the prediction of unobserved variations and has shown an accuracy of over 90% on the entire network of E.Coli along with published experimental data. Notably, such genome-wide predictions can be computed in a few seconds.
From the application perspective, the distinguishing novel features of our approach are as follows: (i) it is fully automated, (ii) it is highly efficient, (iii) it deals with large-scale systems in a global way, (iv) it detects existing inconsistencies between networks and datasets, (v) it diagnoses inconsistencies by isolating their source, (vi) it offers a flexible concept of repair to overcome inconsistencies in biological networks, and finally (vii) it enables prediction of unobserved variations (even in the presence of inconsistency).
The efficiency of our approach stems from advanced Boolean Constraint Technology, allowing us to deal with problems consisting of millions of variables. Although the basic tools [1] are implemented in C++ we have improved their accessibility by providing a Python library as well as a corresponding Web service [2]
Our project is a joint effort between the Knowledge Representation and Reasoning group [3] at the University of Potsdam and the SYMBIOSE Team [4] at IRISA and INRIA in Rennes. Our techniques have been developed in strong collaboration with the Max-Planck-Institute for Molecular Plant Physiology in Potsdam within the GoFORSYS Project [5] as well as Institut Cochin, Paris [6]. The members of the group include Sylvain Blachon, Martin Gebser, Carito Guziolowski, Jacques Nicolas, Max Ostrowski, Torsten Schaub, Anne Siegel, Sven Thiele, and Philippe Veber.
Links:
[1] http://en.wikipedia.org/wiki/Answer_set_programming
[2] http://potassco.sourceforge.net
[3] http://www.cs.uni-potsdam.de/ bioasp/sign_consistency.html
[4] http://www.cs.uni-potsdam.de/wv
[5] http://www.irisa.fr/symbiose
[6] http://www.goforsys.org
[7] http://www.cochin.inserm.fr
Please contact:
Torsten Schaub
Universität Potsdam, Germany
E-mail:
Anne Siegel
CNRS/IRISA, Rennes, France
E-mail:
