









by Knut Reinert
Maybe you like Britney Spears. Maybe even her music. Maybe you are a Britney Spears fan working as part of a group on sequence analysis algorithms in computational biology. But am I right in assuming that you don’t like to hear the above song title quoted by your coworkers or programmers when they could have been spending their time doing something productive or creative? If I am right, then you might want to read on because I will tell you how you, or your coworkers can avoid reinventing the wheel or writing a lot of inefficient scripts in sequence analysis.
Next generation sequencing (NGS) is a term coined to describe recent technological advances in DNA sequencing. Next generation sequencing allows us to sequence about 200.000.000.000 base pairs within approximately one week. That’s a two with many zeros. To explain it in different terms, whilst the human genome project spent many years and billions of dollars to sequence about 30 billion base pairs, we can now perform the equivalent amount of work within a day for a handful of dollars.
DNA as mass data has wonderful properties. While the sequencing machines churn out terabytes of data, a single byte of this data can be very important. It can decide whether you have a disease or not, whether the drugs you take do their job or not, whether you live or die. So we have to treat the data carefully. It is important to scientists working in the life sciences. At the same time, however, we need to process it fast and efficiently, after all it fills the racks of terabyte disks quickly.
In order to analyse the ever-growing volume of sequencing data it is essential that scientists in the life sciences and bioinformaticians work closely together with scientists in computer science. This can often be problematic since both communities approach the problems at hand quite differently (see Figure 1). While the the experimentalists have a holistic top-down view on what they want to achieve in a particular analysis, computer scientists usually work on a specific, small part of a larger analysis problem. On the computer science side this often results in highly efficient, but specialized algorithms that may not necessarily reflect the reality of real world data. Efforts from the life sciences side, in contrast, may result in analysis pipelines that compute a solution to the problem but are not state-of-the-art in run time or memory consumption, and hence cannot be applied to the large data volumes. The goal is obviously to use fast implementations of efficient algorithms to be able to cope with the volume of sequence data that NGS can produce. This can be achieved through algorithm libraries that collect efficient implementations of state-of-the-art algorithms, the work of algorithm designers and skilled programmers, and make them available to the data analysts and bioinformaticians. Apart from the obvious benefit of being able to efficiently compute solutions, the use of software libraries also avoids the Britney Spears effect, namely doing many things all over again, because many algorithms needed are already available in the library.
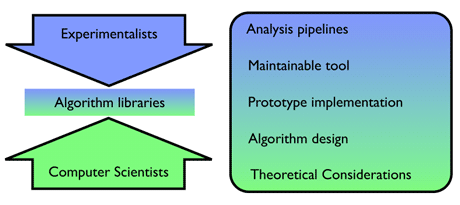
Figure 1: Top-down versus bottom-up approach.
For the past seven years the Algorithmic Bioinformatics group at the FU Berlin has been working on a comprehensive algorithm library for sequence analysis.
The SeqAn project fills the gap between the experimentalists and algorithm designers. SeqAn is a C++ library and has a unique generic design based on:
- the generic programming paradigm
- a new technique for defining type hierarchies called template subclassing
- global interfaces
- metafunctions, which provide constants and dependent types at compile time.
The design of SeqAn differs from common programming practice, in particular SeqAn does not use object-oriented programming. The main consequence of this design choice is that SeqAn implements features like polymorphism at compile time thus avoiding costly run time operations like consulting a lookup table to find the appropriate virtual function, as it is necessary in object-oriented programming. This sets it also apart from other frameworks in the field like Galaxy (http://bitbucket.org/galaxy/galaxy-central/wiki/Home) or BioJava (http://biojava.org/wiki/Main_Page) and BioPerl (http://www.bioperl.org/wiki/Main_Page). SeqAn is intended to embrace high-performance algorithms from the computer science field and to cover a wide range of topics of sequence analysis. It offers a variety of practical state-of-the-art algorithmic components that provide a sound basis for the development of sequence analysis software. These include:
- data types for storing strings, segments of strings and string sets
- functions for all common string manipulation tasks
- data types for storing gapped sequences and alignments in memory and on disk
- algorithms for computing optimal sequence alignments
- algorithms for exact and approximate (multiple) pattern matching
- algorithms for finding common matches and motifs in sequences
- string index data structures
- graph types for many purposes like automata, alignment graphs, or HMMs
- standard algorithms on graphs.
SeqAn has growing user community throughout the EU and US and is actively developed by 4-6 scientists mainly at the FU Berlin. Potential users might be interested in the SeqAn book (see link below). SeqAn is partially supported by the DFG priority program 1307 “Algorithm Engineering”.
Links:
SeqAn project: http://www.seqan.de
http://crcpress.com/product/isbn/9781420076233
Please contact:
Knut Reinert
Algorithmic Bioinformatics, Freie Universität Berlin, Germany
Tel: +49 30 838 75 222
E-mail:
