








by Eric Rivals, Alban Mancheron and Raluca Uricaru
Some bacterial species live within the human body and its immediate environment, and have various effects on humans, such as causing illness or assisting with digestion, while others colonize a range of other ecological niches. The availability of whole genome sequences for many bacteria opens the way to a better understanding of their metabolism and interactions, provided these genome sequences can be compared. In collaboration with biologists and mathematicians, a bioinformatics team at Montpellier Laboratory of Informatics, Robotics, and Microelectronics (LIRMM) has been developing novel tools to improve bacterial genome comparisons.
With the successful sequencing of the entire genome sequence of some species, the 1990s heralded a post-genomic era for biology: for the first time, the complete gene set of a species was amenable to - mostly in silico - investigations. Through comparative genomics approaches, seminal works have, for instance, determined the core gene set across nine bacteria, ie those genes indispensable to the life of any of these species.The availability of whole genome sequences has consequently opened up new research avenues. The current revolution of sequencing techniques provides such an improvement in yield that it brings within reach the sequencing of thousands of bacterial, or even longer, genomes within a few years. However, the exploitation of the information encoded in these sequences requires an increased capacity to compare whole genome sequences. Indeed, even finding the genes within the genome starts by comparing it to that of the evolutionary most closely related species, if available. Today, genome comparisons are necessary in order to survey the biodiversity of genotypes within populations, and to design vaccines .
Given that genomes are DNA sequences evolving under both point mutations, which alter a base at a time, and block rearrangements, ie duplication, translocation, inversion, or deletion of a DNA segment, comparing genomes is formulated as a string algorithm problem.
Two lines of approach have been followed: (i) extend gene sequence alignment algorithms by making them more efficient and coupling them with a procedure to deal with some types of rearrangements, (ii) consider only block rearrangements on a sequence of ordered markers (typically genes), but disregard the DNA sequence. Type two approaches adequately model changes in the gene order, but require well annotated genes and the knowledge of their evolutionary relationships across species, and are unable to report functionally relevant conservation or evolution in non-gene coding DNA regions. Note that handling multiple comparisons makes most of these formulations NP-hard, and, as with most evolutionary questions, benchmarks are missing since evolution occurs once and cannot be replayed. Thus, currently available solutions are time consuming, require fine parameter tuning, deliver results that are hard to visualise and interpret, and furthermore, do not fit the whole spectrum of applications.

Figure 1: Different anchors for whole genome alignment. Alignment of fragments of Brucella suis and B. microtii genomes showing on one hand Maximal Exact Matches (MEM) of length > 5 and a (much longer) Local Alignment. Identical bases are shown in blue background color.
In the framework of the project "Comparisons of Complete Genomes (CoCoGen)" supported by the French National Research Agency, we designed novel algorithms for comparing complete bacterial genomes.
Genome alignment algorithms first search for anchors, which are pairs of matching genomic regions, and chain them to maximize the coverage on both genomes. A chain is an ordered subset of non-overlapping collinear anchors, meaning that the succession of regions appears in the same order on both genomes. Current tools use pairs of short exact or approximate matches as anchors (Mumer, Mauve, MGA), to be sure to find secure anchors in quite divergent regions. Instead, we focus on local alignments (LA) found using sensitive spaced seeds filtration. The length of local alignments adapts to the divergence level of compared regions, and moreover, LA are selected and ranked according to their statistical significance (see Figure 1). However, an important disadvantage is that the regions of distinct anchors may overlap within a genome. By adapting a Maximum Independent Set algorithm on trapezoid graphs, we exhibited a chaining algorithm that allows overlaps between anchors that depend proportionnally on the anchor lengths. This improved significantly the coverage obtained with LA, and overcomes the overlap created by prevalent genomic structures like tandem repeats.
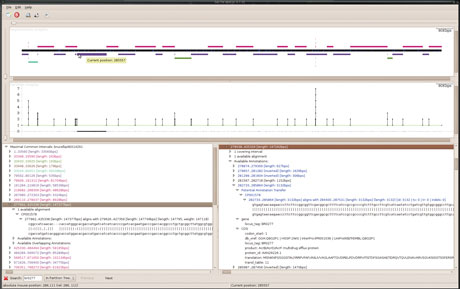
Figure 2: Output of QOD, when used to compare pairwise Brucella suis and B. microtii genomes. The top diagram represents B. suis genome as a central horizontal black and blue bar, and each other colored horizontal bar depicts a maximal region of similarity with B. microtii genome. The high genome coverage by such bars indicates the proximity of the two species. Lower parts of the window allows to browse the segmentation together with intersecting annotations of the target genome.
Unravelling the pathogenicity mechanisms of some bacterial strains requires knowledge of which genomic regions encode the corresponding genes compared to less virulent or non-pathogenic strains. These genes may be relevant in the process of diagnosis, prognosis, and treatment of disease. For instance, a recent approach to vaccine design relies strongly on this type of genome comparison. As many applications of comparative genomics aim at finding such distinguishing regions between strains or species, we conceived methods that can directly pinpoint such regions. In existing algorithms, genome comparison is formulated as a maximization problem and used heuristics tend to incomporate unreliably aligned region pairs in the final output. Consequently, distinguishing regions may be missed due to misalignment, since an anchor can be found by chance in or around such regions. We propose another genome segmentation algorithm that automatically partitions the target genome into regions that are shared or not shared with k reference genomes (see software QOD in Figure 2). Performing several comparisons with a distinct set of reference genomes allows us to rapidly determine which genomic regions distinguish a subset of pathogenic strains/species.
Application of our methods to the comparison of multiple bacterial strains that are pathogenic for ruminants has enabled discovery of the new genes that were overlooked by several previous genome annotation projects. It has also facilitated identification of a gene whose sequence varies among strains, and was known to code for a protein that generates important immunological reactions in related bacterial species (the human and dog pendant pathogens).
Link:
http://www.lirmm.fr/~rivals/CoCoGEN/
Please contact:
Eric Rivals
The Montpellier Laboratory of Informatics, Robotics, and Microelectronics (LIRMM), France
E-mail: