









by Marco Aldinucci, Andrea Bracciali and Pietro Lio'
The human immune system fights pathogens using an articulated set of strategies whose function is to maintain in health the organism. A large effort to formally model such a complex system using a computational approach is currently underway, with the goal of developing a discipline for engineering "synthetic" immune responses. This requires the integration of a range of analysis techniques developed for formally reasoning about the behaviour of complex dynamical systems. Furthermore, a novel class of software tools has to be developed, capable of efficiently analysing these systems on widely accessible computing platforms, such as commodity multi-core architectures.
Computational approaches to immunology represent an important area of systems biology where both multi-scale and time and spatial dynamics play important roles. In order to explore how different modelling and computational techniques can be better integrated to support in silico experiments, a collaboration amongst researchers of Italian and British institutions has been established. In the long term we anticipate that this will lead to the engineering of synthetic immune responses.
In silico experiments involve the reproduction of the dynamics occurring between the immune system and pathogens. These models can be used to simulate the mechanisms and the emerging behaviour of the system, to test new hypotheses and to predict their effects, hence, they will need to be able to measure, analyse and formally reason about the system behaviour. By means of such a virtual lab one can try to design novel "synthetic" responses and drug treatments that may then be implemented in the organic world.
We are exploring the feasibility of combining two research trends, which will certainly be further developed in the near future. The first trend draws from formal methods, ie a set of description techniques that allows qualitative and quantitative aspects of system behaviour to be described and formally analysed. Generally, models consist of populations of agents/entities, or aggregate abstractions of them, eg viruses or lymphocytes. These play a part in the economy of the whole system by means of the possible behaviour they exhibit. The overall behaviour of the system emerges from the interaction between agents and can be observed by means of simulations that account for either probabilistic or averaged evolutions from given initial conditions. Also, the system can be observed in its transient or equilibrium dynamics, and its properties, when precisely expressed in a formal language, can be verified (model checked) against the system behaviour. In particular the formal analysis can be tightly coupled with stochastic simulations in order to improve the information obtained from the simulation results. These approaches, particularly the stochastic ones under certain hypotheses, are extremely computationally expensive.
The second trend aims to enhance the precision and effectiveness of these virtual labs. A good opportunity to achieve this is provided by the recent design shift towards multi-core architectures that make high computational power diffusely available. To exploit this power, however, software tools need to be redesigned to match the new architectures, which may suffer from inefficiencies of the shared memory subsystem due to poor memory access hot-spots and caching behaviour. Within the FastFlow programming framework [1], which provides a cache-aware, lock-free approach to multi-threading, we have developed StochKit-FF (see application page at [1]), an efficient parallel version of StochKit, a reference stochastic simulation tool-kit.
In our reference scenario we use aspects of the HIV dynamics within a patient, as a model infectious disease. Many different viral strains contribute to infection progression; they use different cell receptors (CCR5 and CXCR4) and consequently several anti-HIV therapies target these receptors. Progression of the disease (AIDS) is dependent on interactions between viruses and cells, the high mutation rate of viruses, the immune response of individuals and the interaction between drugs and infection dynamics. These phenomena are inherently stochastic, subject both to intrinsic and extrinsic noises. Some simulation results are shown in Figure 1 and Figure 2.
![Figure 1: A single stochastic simulation over the [1-1000] day interval. Levels of cells from the immune system during HIV infection are shown on the y-axis. Importantly, the decay of T below about 200 represents the passage to AIDS. This emerges at about day 500.](/images/stories/EN82/lio1.png)
Figure 1: A single stochastic simulation over the [1-1000] day interval. Levels of cells from the immune system during HIV infection are shown on the y-axis. Importantly, the decay of T below about 200 represents the passage to AIDS. This emerges at about day 500.
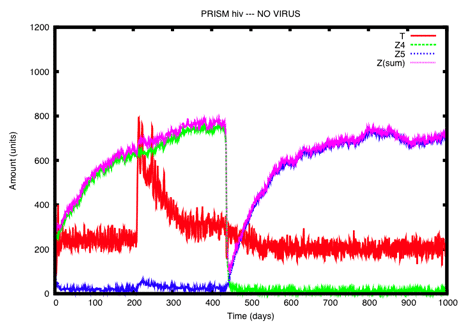
Figure 2: The same scenario as that represented in Figure 1 with the addition of a treatment period starting at about day 200. The effect of the treatment can be “visually” appreciated in terms of an higher amount of T (and Z5) during the treatment period, with respect to the same time interval in Figure 1.
StockKit-FF allows us to run multiple concurrent stochastic simulations efficiently. Results based on a large number of simulations are much more informative than a few probabilistic outcomes. Since the cost of saving comprehensive information about each simulation can become prohibitive, StochKit-FF allows the on-line parallel reduction of solution trajectories from different simulation runs by way of user-defined statistical estimators, such as average and variance. The relative speed of StockKit-FF with respect original StockKit for the HIV model is reported in Figure 3.
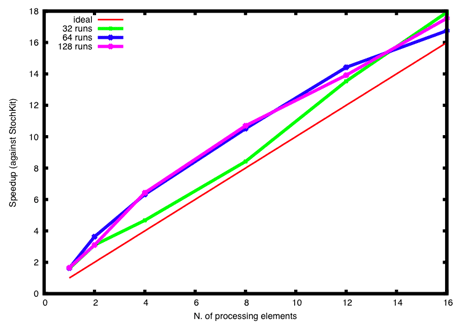
Figure 3: Speedup of StockKit-FF against original StochKit for 32, 64, and 128 runs of the HIV stochastic simulation. StockKit-FF runs are concurrently executed; their outputs are reduced by way of the average and variance functions. StockKit-FF exhibits a super-linear speedup, ie it is always more than n-fold faster than StockKit when running on a n-core platform.
Furthermore, we are quantitatively assessing different scenarios regarding the immune system response. Probabilistic model checking, for instance, can provide a greater understanding of infection models, by shifting the attention from an informative, but empirical, analysis of the graphs produced by simulations towards more precise quantitative interpretations. For instance, this technique can allow us to determine the probability of immune system failure with or without a support therapy over a given time interval. Beyond the visual evidence, it is possible to determine that such a probability for the cases in Figure 1 and Figure 2 is 0.377 vs. 0.855 (results obtained by the PRISM model checker [2], see [3] for details).
Currently, we are investigating more structured ways of collecting information from multiple runs and the possibility of exploiting such multiple runs as approximated model knowledge for model checking purposes. Efficient simulations will make possible an effective investigation of relevant characteristics of the virus attack and the immune defence, while the formal analysis will provide the key tool to identify best anti-viral drug therapy.
This project is a British-Italian collaboration (Universities of Cambridge, Torino and Stirling, ISTI-CNR), partially supported by HPC-Europe, EMBO and the CNR project RSTL-XXL.
Links:
[1] http://mc-fastflow.sourceforge.net
[2] http://www.prismmodelchecker.org/
[3] http://www.biomedcentral.com/1471-2105/11/S1/S67
Please contact:
Andrea Bracciali
ISTI-CNR, Italy and
Computing Science and Mathematics,
University of Stirling, UK
E-mail:
Pietro Lio'
Computer Laboratory
University of Cambridge
E-mail:
