









by Eric Rivals, Nicolas Philippe, Mikael Salson, Martine Leonard, Thérèse Commes and Thierry Lecroq
With High Throughput Sequencing (HTS) technologies, biology is experiencing a sequence data deluge. A single sequencing experiment currently yields 100 million short sequences, or reads, the analysis of which demands efficient and scalable sequence analysis algorithms. Diverse kinds of applications repeatedly need to query the sequence collection for the occurrence positions of a subword. Time can be saved by building an index of all subwords present in the sequences before performing huge numbers of queries. However, both the scalability and the memory requirement of the chosen data structure must suit the data volume. Here, we introduce a novel indexing data structure, called Gk arrays, and related algorithms that improve on classical indexes and state of the art hash tables.
Biology and its applications in other life sciences, from medicine to agronomy or ecology, is increasingly becoming a computational, data-driven science, as testified by the launch of the Giga Science journal (http://www.gigasciencejournal.com)). In particular, the advent and rapid spread of High Throughput Sequencing (HTS) technologies (also called Next Generation Sequencing) has revolutionized how research questions are addressed and solved. To assess the biodiversity of an area, for instance, instead of patiently determining species in the field, the DNA of virtually all species present in collected environmental samples (soil, water, ice, etc.) can be sequenced in a single run of a metagenomic experiment. The raw output consists of up to several hundred million short sequencing reads (eg from 30 to 120 nucleotides with an Illumina sequencer). These reads are binned into classes corresponding to species, which allow to reliable estimation of their number and relative abundance. This becomes a computational question.
In other, genome-wide, applications, HTS serve to sequence new genomes, to catalogue active genes in a tissue, and soon in a cell, to survey epigenetic modifications that alter our genome, to search for molecular markers of diseases in a patient sample. In each case, the read analysis takes place in the computer, and users face scalability issues. The major bottleneck is memory consumption. To illustrate the scale, currently sequences accumulate at a faster rate than the Moore law, and large sequencing centres have outputs of gigabases a day, so large that even network transfer becomes problematic.
Let us take an example. Consider the problem of assembling a genome from a huge collection of reads. Because sequencing is error prone and the sequenced DNA vary between cells, the read sequences are compared pairwise to determine regions of approximate matches. To make it feasible, potentially matching regions between any read pair are selected on the presence of identical subwords of a given length k (k-mer). For the sake of efficiency, it is advantageous, if not compulsory, to index once for all the positions of all distinct k-mers in the reads. Once constructed, the index data structure is kept in main memory and repeatedly accessed to answer queries like ‘given a k-mer, get the reads containing this k-mer (once/at least once)’. The question of indexing k-mers or subwords has long been addressed for large texts, however classical solutions like the generalized suffix tree, or suffix array require too much memory for a read collection. Even state of the art implementations of sparse hash tables (Google sparse hash) hit their limits with such data volumes.
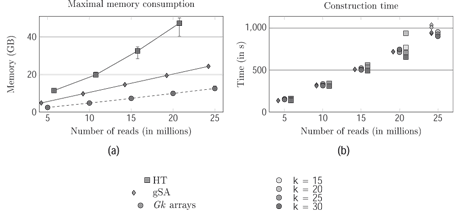
Figure 1: Comparison of Gk arrays with a generalised Suffix Array (gSA) and a Hash Table solutions on the construction time and memory usage.
To address the increasing demand for read indexing, we have developed a compact and efficient data structure, dubbed Gk arrays, that is specialized for indexing huge collections of short reads (the term ‘collection’, rather than ‘set’, underlines that the same read sequence can occur multiple times in the input). An in-depth evaluation has shown that Gk arrays, can be constructed in a time similar to the best hash tables, but outperform all concurrent solutions in term of memory usage (Figure 1). The Gk arrays combine three arrays: one for storing the sorted positions where true k-mers start, an inverted array that allows finding the rank of any k-mer from a position in a read, and a smaller array that records the intervals of positions of each distinct k-mer in sorted order. Although reads are concatenated for construction, Gk arrays avoid storing the positions of (artificial) k-mers that overlap two adjacent reads. For instance, the query for counting the read containing an existing k-mer takes constant time. Several types of queries have been implemented and Gk arrays accommodate fixed as well as variable length reads. Gk arrays are packaged in an independent C++ library with a simple and easy to use programming interface (http://www.atgc-montpellier.fr/ngs/). They are currently exploited in a read mapping and RNA-sequencing analysis program; their scalability, efficiency, and versatility made them adequate for read error correction, read classification, k-mer counting in assembly program, or other HTS applications. Gk arrays can be seen as an indexing layer that is accessed by higher level applications. Future developments are planned to devise direct construction algorithms, or a compressed version of Gk arrays that, like other index structures, stores only some sampled positions and reconstruct the others at runtime, hence enabling the user to control the balance between speed and memory usage.
Gk arrays library is available on the ATGC bioinformatics platform in Montpellier: http://www.atgc-montpellier.fr/gkarrays
Link:
Gk arrays library:
http://www.atgc-montpellier.fr/gkarrays
Please contact:
Eric Rivals
LIRMM, CNRS, Univ. Montpellier II, France
E-mail:
http://www.lirmm.fr/~rivals
