









by Marco Hülsmann, Thorsten Köddermann, and Dirk Reith
The Fraunhofer Institute for Algorithms and Scientific Computing (SCAI) has developed a software tool for the automated parameterization of force fields for molecular simulations using efficient gradient-based algorithms. This tool, combined with well-established simulation techniques, can quantitatively determine many physicochemical properties for given compounds.
In the field of process engineering performed by chemical and pharmaceutical industries, physicochemical knowledge like vapor-liquid and liquid-liquid equilibria, or understanding the relationship between molecular structure and macroscopic properties, is essential. In the past two decades, the availability of increasingly powerful computers has opened routes towards obtaining various structural, dynamic and thermodynamic properties from relatively inexpensive computational models.
Molecular simulations relate macroscopic phenomena to their roots in molecular interactions. The practical applicability of these simulations in process engineering requires the construction of appropriate molecular models for a wide range of chemicals. In this context, the Fraunhofer Institute SCAI has developed the GRadient-based Optimization Workflow (GROW) software tool, which makes possible the fast and reliable development of models. For their customers, SCAI is now able to calculate quantitative physicochemical properties of chemicals and their mixtures at a wide range of temperatures and pressures. Furthermore, it is possible to explore in silico how changing the molecular structure influences chemical properties – thus guiding future experiments. With GROW, SCAI is well prepared to assist industry in its endeavour to engineer new chemical substances.
Molecular simulations are widely used to support the development process of new materials. Through simulations researchers are able to predict qualitative trends quite well. However, the key to quantitative property predictions is the accuracy of a simulation's foundation, the force field. While the equation's functional form is usually straightforward, the force field parameterization is often tedious. Manual adjustment and optimization is at best extremely time-consuming. In our pursuit to create tailor-made models for specific investigations in a timely fashion, an automated parameterization scheme is therefore essential.
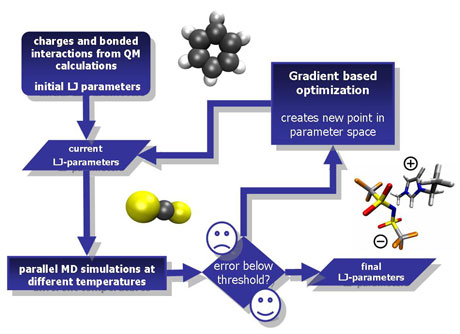
Figure 1: The optimization workflow: physicochemical properties are calculated from an initial guess of the force field parameters. If the calculated properties do not agree sufficiently well with the experimental data, a gradient-based optimization is performed. This process is iteratively performed until a stopping criterion is fulfilled and the final parameters are found.
While intramolecular model parameters and partial atomic charges can be obtained from quantum mechanical calculations, the determination of intermolecular parameters, eg Lennard-Jones parameters, is much more challenging. The aim is to fit these parameters to a selection of experimental properties, such as density, enthalpy of vaporization, self-diffusion coefficient, vapur pressure and reorientation time. The resulting force fields can be used to reliably predict other properties of pure components as well as heterogeneous solutions.
GROW is a program tool kit that facilitates the gradient-based numerical optimization of force field parameters. Its components include various efficient optimization algorithms, analysis scripts and I/O handling. At the core of GROW is a gradient-based minimization of a quadratic loss function. The loss function is taken between experimental and simulated physicochemical properties at various temperatures. Specifically, the minimization algorithms implemented in GROW are the Steepest Descent, Newton, Conjugate Gradient and Trust Region methods.
Through an automated iterative process, in a parallel computer environment, the optimized solution is realized and new molecular models are developed. GROW can be coupled to various standard simulation engines, making it a powerful companion for many application fields.
GROW is flexible with regard to the simulation engine, the molecules of interest and the physicochemical properties used for target fitting. Hence, it can be broadly applied to scientifically and industrially relevant problems.
Experimental target values can be typically matched within a few percent over the considered temperature range for common properties; for example, the error is approximately 0.5-1% for density and 2-3% for enthalpy of vaporization and self-diffusion coefficients. This has been illustrated for carbon disulfide, benzene, phosgene and ionic liquids, and in general, fewer than ten optimization steps are required to obtain results in this order of magnitude. The physicochemical properties that can be simulated include shear viscosity, self-diffusion coefficients, electrical and thermal conductivity, density, compressibility, enthalpy of vaporization, vapor pressure, octanol-water partition coefficient, solubility of compounds, activity coefficient and surface tension.
In a further step, an optimized force field obtained by GROW for a specific compound can be transferred to chemically modified structures thereof. This enables one to study the influence of the molecular structure on certain properties.
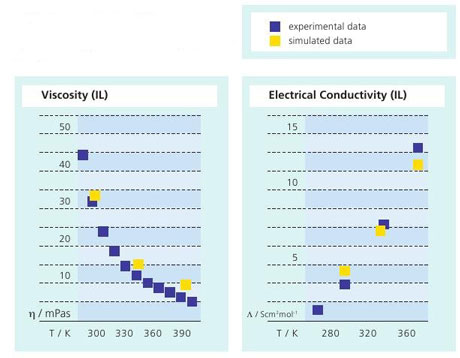
Figure 2: Simulated viscosity and electrical conductivity of an ionic liquid as a function of temperature compared to experimental data. The simulated results are in the range of the experimental error.
To summarize, one of the main problems in the field of computational chemistry engineering is the optimization of intermolecular force field parameters. With the development of GROW, the Fraunhofer Institute SCAI has made an important step toward solving this problem in an efficient way.
Please contact:
Dirk Reith
Fraunhofer Institute for Algorithms and Scientific Computing (SCAI), Germany
Tel: +49 2241 14 2746
E-mail:
